




 该文提供了一种方法,使用Python和R脚本来整理10x单细胞转录组数据的多个文件。脚本根据GSM号和样本名自动创建文件夹并将相关文件归类,方便生物信息学分析。
该文提供了一种方法,使用Python和R脚本来整理10x单细胞转录组数据的多个文件。脚本根据GSM号和样本名自动创建文件夹并将相关文件归类,方便生物信息学分析。
多个10x单细胞转录组每个样品的3个文件如何归纳到同一个文件夹里面 (看到曾老师收费不能白嫖 我就写个脚本)
首先 我下载了一个10x的文件集合哈
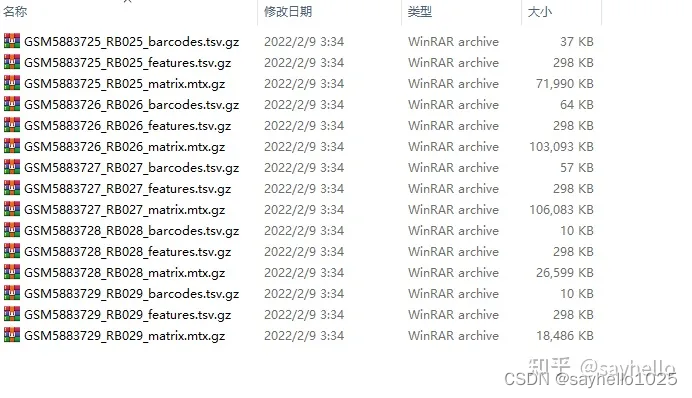
我的理想是整成这个样
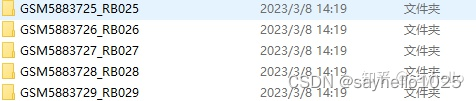
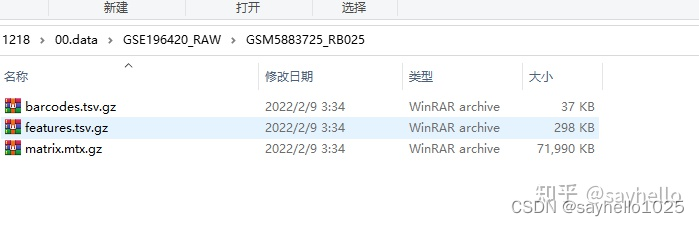
打开后是这个样式
如何实现呢我提供了py的脚本,让chatgpt注释下给大家看
# 导入必要的模块
import os
import re
import shutil
# 获取当前目录下所有以'.gz'结尾的文件列表
files = [f for f in os.listdir() if re.match('.*\.gz$', f)]
# 遍历每个文件
for i in files:
# 从文件名中提取出GSM号
GSM = re.findall('(?<=GSM)\\d+', i)[0]
# 从文件名中提取出样本名
sample = re.findall('(?<=GSM\\d{7}_)[^_]+', i)[0]
# 生成新的文件夹名字
filename = f'GSM{GSM}_{sample}'
# 如果文件夹不存在,则创建文件夹
if not os.path.exists(filename):
os.mkdir(filename)
# 根据GSM号生成文件名的正则表达式
filepattern = f'GSM{GSM}.*\.gz$'
# 获取所有以GSM号开头,以'.gz'结尾的文件列表
files1 = [f1 for f1 in os.listdir() if re.match(filepattern, f1)]
# 遍历每个文件,将文件移动到对应的文件夹中
for file in files1:
# 从文件名中提取出新的文件名
new_filename = file.split('_')[2]
# 将文件移动到对应的文件夹中
shutil.move(file, os.path.join(filename, new_filename))
考虑到生物信息的同学们用R 我也写了个R 的版本
library(stringr)
files <- list.files(pattern = ".gz$")
for (i in files) {
GSM <- str_extract(i, "(?<=GSM)\\d+")
sample <- str_extract(i, "(?<=GSM\\d{7}_)[[:alnum:]]+")
filename <- paste0('GSM',GSM,'_',sample)
if (!dir.exists(filename)){
dir.create(filename)
}
filepattern <- paste0('GSM',GSM,'.*\\.gz$')
files1 <- list.files(pattern = filepattern)
# 移动文件
lapply(files1, function(file) {
new_filename <- strsplit(file,'_',)[[1]][3]
file.rename(file,
paste0(filename,'/',new_filename))
})
}



















 9506
9506

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









