







Cu单晶结构弛豫与晶格常数计算
使用 LAMMPS 进行铜(Cu)单晶的结构弛豫和晶格常数计算时,需要基于分子动力学(MD)或能量最小化方法,结合合适的原子间势函数(如嵌入原子法势 EAM)。以下是详细的操作步骤和示例脚本:
1. 建模与初始结构
铜的晶体结构为面心立方(FCC),晶格常数实验值约为 3.615 Å。(这里设置为 5 Å,变化较为明显)
在 LAMMPS 中可通过 lattice 命令生成初始结构:
# 基础设置
units metal
boundary p p p
atom_style atomic
# 创建 FCC 晶格
lattice fcc 5
region box block 0 4 0 4 0 4 #四个晶胞
create_box 1 box
create_atoms 1 box
#输出初始结构
write_data initial_structure.data # 指定输出文件名
2. 势函数选择
铜的模拟常用 EAM(嵌入原子法)势,例如:
Cu_u3.eam:Cu_u3.eam.txt
- 由 M.I. Baskes 等人开发,基于早期的 EAM 模型。
- 拟合的数据集相对较小,主要针对纯铜的体相性质(如晶格常数、弹性常数等)。
- 适用于简单的体相模拟,但在表面、缺陷或高温条件下的表现可能不够精确。
Cu_u6.eam:Cu_u6.eam.txt
- 由 Y. Mishin 等人开发,基于更广泛的实验和第一性原理数据。
- 拟合的数据集更大,包括体相、表面、缺陷、液态和高温性质。
- 适用于更复杂的模拟场景,如表面重构、晶界、位错和熔化行为。
在 LAMMPS 中加载势函数:
pair_style eam
pair_coeff * * Cu_u6.eam Cu
# 设置邻居列表
neighbor 2.0 bin
neigh_modify delay 10 check yes
3. 结构弛豫(能量最小化)
通过能量最小化优化原子位置和晶格常数:
# 输出热力学信息
thermo 1
thermo_style custom step temp pe etotal press vol lx ly
#结构弛豫(能量最小化)
min_style cg
minimize 1.0e-25 1.0e-25 10000 10000
4. 晶格常数优化
若要自动优化晶格常数,需启用 体积弛豫(通过 fix box/relax):
# 盒子的体积弛豫 (各向同性)
fix 1 all box/relax iso 0.0 vmax 0.001
min_style cg
minimize 1.0e-25 1.0e-25 10000 10000
iso 0.0表示三轴等压弛豫,vmax 0.001控制体积变化步长。- 优化后的晶格常数可通过输出文件中的
lx,ly,lz读取(FCC结构中三者相等)。
注:npt 与 box/relax 的区别
| 特性 | fix box/relax | fix npt |
|---|---|---|
| 功能 | 调整盒子尺寸以最小化能量或应力 | 控制温度和压力,模拟恒温恒压行为 |
| 适用阶段 | 能量最小化 | 分子动力学模拟 |
| 控制温度 | 不支持 | 支持 |
| 控制压力 | 不支持 | 支持 |
| 盒子变化方式 | 各向同性或各向异性 | 各向同性或各向异性 |
| 典型应用 | 消除初始结构的应力 | 模拟体系在指定温度和压力下的行为 |
5. 完整示例脚本
# 基础设置
units metal
boundary p p p
atom_style atomic
# 创建 FCC 晶格
lattice fcc 5
region box block 0 4 0 4 0 4 #四个晶胞
create_box 1 box
create_atoms 1 box
#输出初始结构
write_data initial_structure.data # 指定输出文件名
#势函数选择
pair_style eam
pair_coeff * * Cu_u6.eam
# 设置邻居列表
neighbor 2.0 bin
neigh_modify delay 10 check yes
# 输出热力学信息
thermo 1
thermo_style custom step temp pe etotal press vol lx ly
#结构弛豫(能量最小化)
min_style cg
minimize 1.0e-25 1.0e-25 10000 10000
# 盒子的体积弛豫 (各向同性)
fix 1 all box/relax iso 0.0 vmax 0.001
min_style cg
minimize 1.0e-25 1.0e-25 10000 10000
#输出最终的晶格常数
variable a equal lx/4
print "Optimized lattice constant a = ${a} Angstrom"
# 输出弛豫后的结构
write_data Cu_relaxed.data
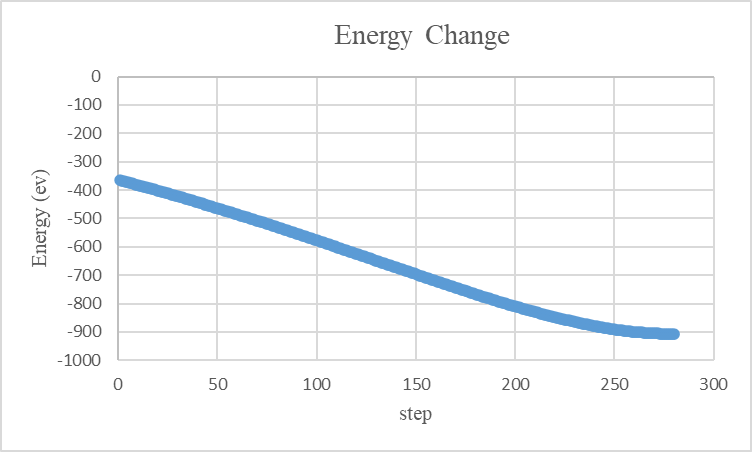
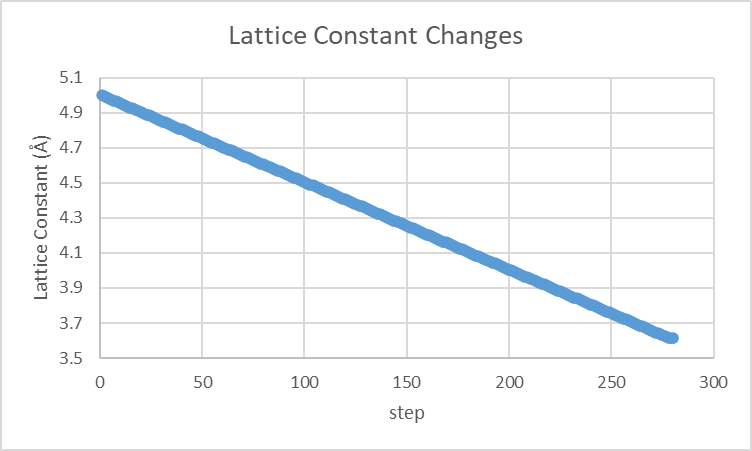
6. 结果分析
- 输出日志:检查
thermo输出中的lx,ly,lz,计算晶格常数的变化。 - 能量收敛:确保
Potential energy (pe)不再显著变化。 - 实验对比:实验值约为 3.615 Å,不同势函数可能导致 ±0.02 Å 偏差。晶格常数计算结果为3.61500011805211 Å。
7. 注意事项
- 势函数选择:务必使用已验证的 Cu 势函数(可从 NIST 或 LAMMPS官网获取)。
- 边界条件:根据体系大小选择合适的周期性边界条件(
boundary p p p)。 - 并行加速:使用多核并行计算加快优化速度(
mpirun -np 4 lmp_mpi -in input.lammps)。
本博客致力于分享知识与信息,部分内容可能引用了网络资源。我会尽力注明作者或来源。如果您认为本博客的内容存在侵权现象(包括但不限于版权、商标权等),请立即与我联系。



















 2844
2844

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











