







 介绍使用R包micrntwk进行微生物群落网络构建的方法,包括计算相关系数、筛选变量、网络拓扑特征分析及可视化。
介绍使用R包micrntwk进行微生物群落网络构建的方法,包括计算相关系数、筛选变量、网络拓扑特征分析及可视化。
以Hmisc包的rcorr函数计算spearman相关系数或minerva包的mine函数计算变量间的连接强度.根据所给阈值筛选变量,将变量之间的关系矩阵转为igraph包的对象,并计算网络拓扑特征。特别的,以qgraph包的qgraph.layout.fruchtermanreingold函数获取网络节点和边的坐标,同时根据用户输入的模块参数,计算每一个模块所含节点数目占总节点的百分比,最后使用ggplot2绘图. 将这些过程总结、写成micrntwk包,安装方式:
##国内用户推荐用方式1
##方式1
remotes::install_git("https://gitee.com/wqssf102/micrntwk", dependencies = TRUE)
##方式2
devtools::install_github("wusu102/micrntwk")
加载包:
library(data.table)
library(tidyr)
library(micrntwk)
library(ggplot2)
library(ggh4x)
以calcor函数计算变量之间的关系系数,可以查看calcor函数的帮助文档:
# ?calcor
# codt 数据框矩阵,一般为物种矩阵
#
# r_th 筛选的R值阈值
#
# p_th 筛选的P值阈值,当type为mine时,无P值
#
# type 可选"cor"或"mine",前者为spearman,后者为最大信息系数
#
# n.cores 当type为mine时起作用,即多线程计算的线程数目
#
# group 若想分组计算变量的关系,给出分组信息即可(为2列,列名分别为sampleid和group)
cordt <- fread("genus.txt",header = T,sep = "\t",data.table = F)
cordt <- separate(cordt,col=taxonomy,into = c("k","p","c","o","f","g"),sep = "[|]")
cordt <- cordt[,!(names(cordt)%in%c("k","p","c","o","f"))]
row.names(cordt) <- cordt[,which(names(cordt)=="g")]
cordt <- cordt[,-1]
##将含有unclassified的行删掉
cordt <- cordt[stringr::str_detect(row.names(cordt),"unclassified",negate = TRUE),]
cordt <- as.data.frame(t(cordt))
crores <- calcor(codt = cordt,group = NULL,type = "cor",r_th = 0.6,p_th = 0.05,n.cores = 4)
crores里包含了3个结果:
gg:igraph对象的变量关系,即网络图;
corgg:变量之间关系的长型数据;
modularity:网络图的模块化系数;
以上结果可用$提取,如crores$modularity.
使用get_node_edg函数获得节点和边的坐标位置,及x和y
ndedinf <- get_node_edg(ggdt = crores,nc = FALSE)
ndedinf里包含了节点和边的坐标信息,可用$提取,如ndedinf$node_inf
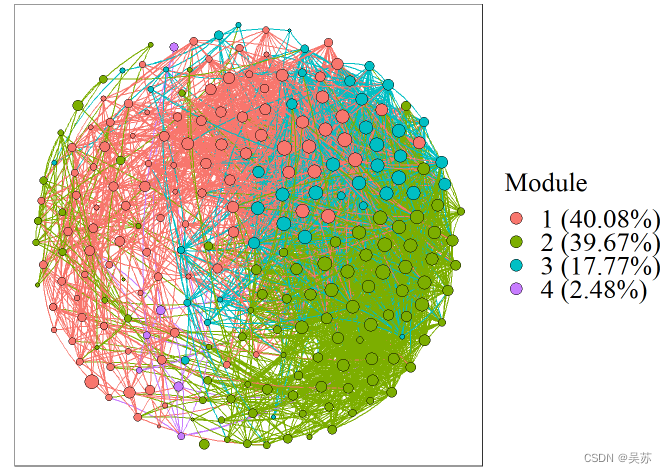
##如下,V1、V2为节点的X、Y坐标,md_name为节点所属模块,mdper为当前模块所有节点所占网络总节点的百分比.
##这里可以看到每个模块包含了哪些节点(即物种或ASV),若要提取某个模块的信息,那就相当容易了
head(ndedinf$node_inf)
V1 V2 node_name Degree md_name mdper
1 102.29885 -79.33870 g__Sphingomonas 32 2 39.67
2 145.45098 -47.63305 g__Rubrobacter 49 2 39.67
3 25.09176 141.72227 g__ADurb.Bin063-1 36 1 40.08
4 72.39232 263.61333 g__uncultured__f__Pirellulaceae 5 3 17.77
5 -150.28829 25.32369 g__uncultured__f__Diplorickettsiaceae 11 2 39.67
6 -173.20600 65.51386 g__Aquicella 13 2 39.67
提取子网络
当crores含有group时,使用
get_mult_sb_nt(ggdt = crores,asvdt = cordt)
当不含group时,使用
get_single_sb_nt(gg = crores$gg,asvdt = as.data.frame(t(cordt)))
使用net_pro_sub函数计算网络拓扑特征.
sbnt <- get_single_sb_nt(gg = crores$gg,asvdt = as.data.frame(t(cordt)))
ntpr <- net_pro_sub(netlist = sbnt)
ntpr中包含子网络的拓扑参数,以inv开头的指标,是指去掉inv之后剩下的指标名的倒数,如inv_average.path.length就是average.path.length(平均最短路径)的倒数.这里取倒数
的目的为使所有指标都是“朝着同一方向”,方便描述网络的复杂性.
可以比较拓扑特征的差异,这时候需要提供分组信息.
grpdt <- read.csv("grp.csv",header = TRUE,row.names = 1)
head(grpdt)
## sampleid group
## site1 site1 AA
## site2 site2 AA
## site3 site3 AA
## site4 site4 AA
## site5 site5 AA
## site6 site6 AA
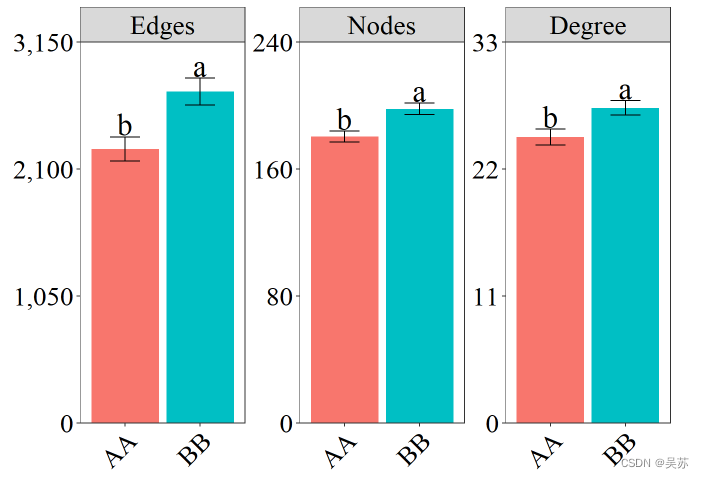
这里只比较3个拓扑参数:
ntprodt <- ntpr
ntprodt <- ntprodt[1:3]
names(ntprodt) <- c("Edges","Nodes","Degree")
dfres <- mydffc(mydfdt = ntprodt,grp = grpdt,mymethod = "LSD",padj = "fdr")###根据自己需要更改mymethod的内容
errbartype <- "se"
if(errbartype=="se"){
errdt <- data.frame(x=dfres$dtmean+dfres$ste,y=dfres$dtmean-dfres$ste)
}else{
errdt <- data.frame(x=dfres$dtmean+dfres$std,y=dfres$dtmean-dfres$std)
}
ggplot(dfres,aes(dtgrp,dtmean,fill=dtgrp)) +
geom_bar(stat="identity",show.legend = FALSE)+
geom_text(aes(x = dtgrp,y = errdt$x,label = dflab),
size = 8,color = "black",vjust=-0.1,position = position_dodge(0.9),family="serif")+
geom_errorbar(data=dfres,aes(x=dtgrp,ymin = errdt$y, ymax = errdt$x),width = 0.4)+
facet_wrap(.~variable,ncol =4,scales = "free_y")+
labs(x=NULL,y=NULL)+
# scale_fill_manual(values = c("#7570B3","#1B9177","#D95F02","#E7298A"))+####根据实际情况提供颜色,也可默认,将此行代码注释掉即可
theme_bw()+
theme(panel.grid = element_blank(),
axis.title = element_text(size = 20,colour = "black"))+
theme(axis.text.y = element_text(colour = "black",size = 20),
axis.text.x = element_text(colour = "black",size = 20,angle = 45,hjust = 1))+
theme(legend.title = element_text(size = 20,colour = "black"),
legend.text = element_text(size = 20,colour = "black"),
strip.text = element_text(size = 20,colour = "black"),
text = element_text(family = "serif"))+##################################以下代码是调整y坐标刻度信息
facetted_pos_scales(y= myf(dfres = dfres,tklb = "variable",fcvar="variable",
numvar = "dtmean",
tknm = 3,###显示多少个刻度
type = "dif",###差异分析
nmlb = 0.1,###坐标小数点位数
axisty = "y",####操作对象为y轴
ax = 1,####将所有指标的数值扩大多少倍,方便调轴的数值、美观。针对于微生物丰度、基因丰度等适用
amax =1.1,###数值大于100的指标的坐标扩大多少倍,方便展示误差棒和字母
amid = 1.1,###数值1到10的指标的坐标扩大多少倍,方便展示误差棒和字母
amix = 1.1,###数值10到100的指标的坐标扩大多少倍,方便展示误差棒和字母
aminx = 1,###数值0.01到1的指标的坐标扩大多少倍,方便展示误差棒和字母
amin = 1###数值小于0.01,方便展示误差棒和字母
))
绘制网络图,可使用plot_net函数,包含参数为:
ex_edg 是否展示网络图的边
per_modules 将节点个数占比低于所给阈值(如30%)的模块统一归类
ex_sp_num 展示多少个物种,若不想展示,设置为0即可
grpnm_position 当含有多个组的网路图时,该参数课控制组名显示的位置,"ld", "lt", "rd", "rt"分别为左下、左上、右下、右上
grpnm_position_num 当含有多个组的网路图时,微调组名位置,如c(1,0.85)为X坐标不变、Y坐标缩小0.85
plot_net函数包含3个结果,为网络图、节点和边的信息,由/$提取,节点和边的信息可提取出来自由绘图:
ptres <- plot_net(netwkinf = ndedinf,ex_edg = T,per_modules = 0,ex_sp_num = 0,point_size = "dg",grpnm_position = "ld")
ptres$plt
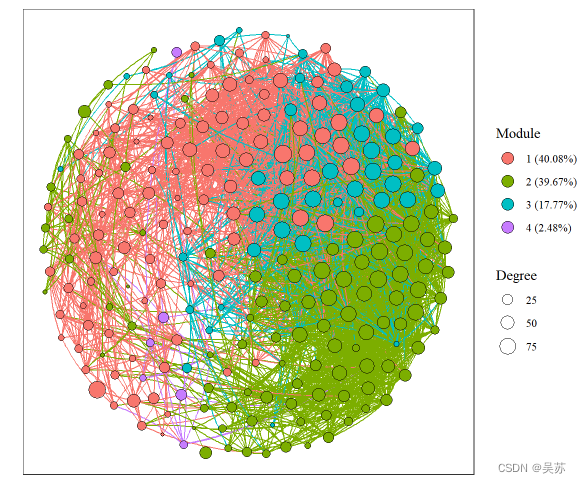
也可以提取边和节点的信息,使用ggplot2绘图
nddt <- ptres$xyinf_node
edgt <- ptres$xyinf_edge
####下面的图没有展示边
ggplot()+
geom_curve(data = edgt,aes(x = V1.x,y=V2.x,
xend=V1.y,yend=V2.y,
color=mdcol),
size = 0.5,curvature = -0.1,show.legend = FALSE)+
geom_point(data = nddt,aes(x=V1,y=V2,size=Degree,fill=factor(mdcol)),shape=21)+
# scale_fill_manual(values = c("1 (56.78%)"="red",......))+####修改模块颜色
labs(x=NULL,y=NULL,fill="Module")+
scale_y_continuous(breaks = NULL)+
scale_x_continuous(breaks = NULL)+
guides(fill=guide_legend(title = "Module",override.aes = list(size=4)))+
theme_bw()+
theme(panel.grid = element_blank(),
strip.text = element_text(size = 20,colour = "black"),
text = element_text(family = "serif"))
分组计算并可视化:
##这里的type我选为mine
grpdt <- read.csv("grp.csv",header = TRUE,row.names = 1)
crores <- calcor(codt = cordt,type = "mine",r_th = 0.6,p_th = 0.05,n.cores = 4,group=grpdt)
ndedinf <- get_node_edg(ggdt = crores,nc = FALSE)
##plot_net包含多个参数,可查看帮助文档
ptres <- plot_net(netwkinf = ndedinf,ex_edg = T,per_modules = 5,ex_sp_num =5,point_size = "dg",nrow = 1,node_text_size = 6,grpnm_position = "ld")
ptres$plt
从几个现成的R包中提取相应的函数计算相关系数、获取网络图中节点、边的坐标,最后使用ggplot2绘图,可方便我们在图中添加信息,比如,使用物种对应的水平、功能给节点着色或形状。
特别的,用户可输入其他算法获得的相关系数矩阵,然后传递给micrntwk包,即可进行拓扑特征计算和完成可视化,只需要将导进来的数据转为igraph包对象并且修改其属性(class)即可,如:
library(igraph)
BFtr1.edge <- read.csv("BFtr1_network.csv")
head(BFtr1.edge)
#Source Target
#ASV_122 ASV_1
#ASV_237 ASV_1
#ASV_264 ASV_1
#ASV_62 ASV_1
BFtr1.net <- graph_from_data_frame(BFtr1.edge[,1:2],directed = FALSE)
haha <- list(gg=BFtr1.net)
class(haha) <- "calcor"
ndedinf <- get_node_edg(gg = haha,nc = FALSE)
ptres <- plot_net(netwkinf = ndedinf,ex_edg = F,per_modules = 0,ex_sp_num = 0,point_size = "dg",grpnm_position="ld")
ptres$plt
提取模块包含的物种
##请注意,这里的函数已经根据模块所包含的物种个数对模块重新命名,
##使得排在前面的模块的物种数最大,如使得模块1的物种数多于模块2
spdata <- get_module_sp(ggdt = crores,spdt = cordt,seed = 123456)
##但这里的模块包含N个物种,一般地,可以使用PCA、PCoA、Z-sore转化等方法,将N个物种的丰度表(即N列数据)转为1列,这1列的数据代表整个模块的物种信息
#提取AA组的module1并做Z-sore转化
mdspneed <- as.data.frame(apply(spdata$AA$module1, 2, function(x){
(x-mean(x))/sd(x)
}))
mdspneed <- as.data.frame(apply(mdspneed, 1, mean))
names(mdspneed) <- "Z_score"
##一个网络图一般包含多个模块,用循环来提取并做Z-sore转化
spdata_A <- spdata$AA
names(spdata_A)##可以看到spdata_A包含了group内容,这里我们不需要group,因此删掉
spdata_A <- spdata_A[!(names(spdata_A)%in%"group")]
##构建一个空的列表,存放每一个模块的结果
mddtlist <- list()
for(i in names(spdata_A)){
ctdt <- spdata_A[[i]]
ctdt <- as.data.frame(apply(spdata$AA[[i]], 2, function(x){##这里提取AA组,根据在自己的实际情况更改
(x-mean(x))/sd(x)
}))
ctdt <- as.data.frame(apply(ctdt, 1, mean))
names(ctdt) <- i
mddtlist[[i]] <- ctdt
}
####将list转为dataframe,按列合并
mddtlist <- do.call(cbind,lapply(mddtlist, data.frame))
head(mddtlist)##查看结果,即可得到每一个模块的结果,接下来读取环因子数据,即可根据自己需要做分析

















 1万+
1万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









