



完整代码:
https://download.youkuaiyun.com/download/weixin_55771290/87944120
摘 要
如今Pubmed文献检索系统上发表的医学文献的数量十分庞大,且数量逐年增加,研究人员如果想人工地去查看找出Pubmed文献里面的知识是绝对不可能,因此,人们转而利用计算机去获取文献里面的知识。
本篇论文介绍了如何借用文本挖掘技术去挖掘出Pubmed文献里面的知识,并且结合了目前文本挖掘技术,讲述了如何实现了一套蛋白质磷酸化修饰的文本信息挖掘系统。
本系统主要应用于挖掘出Pubmed文献里面蛋白质磷酸化的修饰的一些信息,包括被修饰的蛋白质,激酶,修饰位点,以及它们之间的关系。
本文详细叙述了整套系统瀑布流模型的软件过程,首先是需求,然后是设计,再是实现,依次展开。在实现的阶段里面又包含了文本预处理阶段,命名实体识别阶段,实体关系提取阶段,数据可视化阶段,其中着重介绍了文本挖掘技术中两个的关键也是核心阶段的原理:命名实体识别和关系提取。同时也介绍了Abner工具和Rlims-p工具的原理和应用。此外文献数据库的数量庞大,为了提高程序性能和用户体验,于是介绍了几种提高效率,提高用户体验的解决方案,其中有多线程处理,缓存机制,预处理机制。
关键词:文本挖掘;软件工程;Pubmed;多线程
Abstract
Nowadays, The number of published medical literature on Pubmed document retrieval system is very large, growth year after year. It is absolutely impossible to researchers manually discover knowledge among the Pubmed literatures. As a result, researchers turn to use the computer to acquire knowledge inside the literature.
This paper introduce how to acquire knowledge in Pubmed literature by using the technology of text mining and how to implement a text mining system for extracting protein phosphorylation information among Pubmed literature using the current text mining technology. This system is main used for extracting protein phosphorylation information among the Pubmed literatures such as substrate, kinases, sites, and relation among these substances. With the current text mining technology, I
This paper describes the waterfall model of software process about this system. First step is requirement, second step is design, and then implementation, step by step.In the step of implementation, there are four steps as follow: text preprocessing, named entity recognizeation, relationship extracting, Visualization. This paper highlights the principle and application of two very important steps in text mining: named entity recognization, and relationship extracting. At the same time, Abner tools and Rlims-p tools are also introduced in this paper. Moreover, the number of the Pubmed literatures in database is very large, and in order to improve the program performance and user experience,this paper had introduced several method for improve the program performance and user experience such as multithreading, caching, preprocessing.
Keyword: Text mining, Software engineering, Pubmed, Multithreading
第一章 引言
1.1 概述
在近十几年来,计算机领域发展迅猛,个人计算机,终端互联设配也越来越普及,几乎是人人随时随地都能上网,在当今互联网这个巨大的生态圈里面,每年互联网用户产生的数据正以惊人的速度增涨。这些逐年增长的数据中绝大多数的数据都是非结构化的数据,这些数据主要存储的形式都是文本、网页、图片、视频、音频等非结构化的形式。结构化的数据它是经过了一定的存储格式,处理规则加以封装,所以它能够被计算机根据其存储的规则来进行识别,而非结构化的数据比如说文本数据,这些数据没有规则,格式,通常情况下它是不能被计算机直接识别的。当人们想对这些数据进行分析,统计,挖掘的时候,由于其不是结构化的数据,很难通过数据挖掘的技术对其进行处理。人工筛选的方法在这个大数据时代显然是天方夜谭,虽然说人工筛选的方法在上世纪80年代之前都是的确存在的,有需求就有市场这句话放在科研的领域也是行的通的,正因为有对非结构化文本识别和分析的需求,所以研究者们也想出了各种相关的方法。文本挖掘(Text Mining),图像识别,模式识别等一些相关技术就对分析和识别这一些非结构化的数据起到很好的作用。在此篇论文中我们主要讨论一下文本挖掘技术。
文本挖掘(Text Mining)是一个从非结构化文本信息中获取用户感兴趣或者有用的模式的过程[1]。首先它会把非结构化的文本数据通过一定的方法转换成为结构化的数据,然后再从这些结构化的数据中分析提取出我们用户所感兴趣的模式和东西。文本挖掘技术是一项非常复杂的技术,它是一个多学科混杂的一门学科,其中包含有信息技术、机器学习、统计学、数据可视化、数据库技术、数据挖掘[2]。集这么多门学科于一身,它在我们这个大数据时代,信息化时代发挥了重要的作用,也是在信息学科领域中比较热门的学科之一。
在生物医学领域,借助着计算机科学技术也在快速的发展,每年发表在互联网上的生物医学文献资料和研究者们在实验室产生的实验数据也正以惊人的速度增长[3],产生的这些文献资料和实验数据大部分都是以文本的形式存储在计算机设配上面。所以也是非结构化的数据,不能被计算机直接识别。那么我在此篇论文里主要讨论针对文本形式存储的非结构化数据的处理以及信息的提取。
1.2 选题的背景和意义
基因和蛋白质一直都是生物医学工程方面的两大热点词汇,人类从刚开始发现它们到现在一直都在研究它们,并从未停止过对它们的研究和实验。经过科学家们的众多次探索和实验让我们对它们有了一定的了解和认识,并且也享受到了研究成果给人类带来的益处.蛋白质是由氨基酸排列组合生成的,人体氨基酸种类有20种,这20种氨基酸排列组合可以生成成千上万种的蛋白质[4],随着研究者们不断深入研究蛋白质和不断实验,发表在科学文献期刊上的文献数量也随之增加,这个数量正在指数级地增长,如此多的学术文献给研究者们带来了无比多的信息和知识,但是蛋白质的种类如此繁多,有关蛋白质的文献也就越多,这么多的科学文献,研究者是不可能全部阅读完的,这样一来数量如此庞大的科学文献对于研究者们来说是一种浪费,然而研究者们发现他们在阅读每一篇文献时,仅仅只是要获取其中的蛋白质的关键信息,但是他们不得不逐一整片阅读,这大大阻碍了他们的研究效率,所以研究者们迫切需要借用计算机大数据的手段来支持他们完成更深入的探索和研究[5]。他们希望通过计算机的文本挖掘和文本分析来帮他们迅速定位到他们想要得到的蛋白质信息,所以文本挖掘技术迫切需要应用与生物医学文献挖掘上,这也是我本论文的核心研究的方向。
在如此庞大的生物医学文献库当中加之以文本挖掘技术进行适当分析和处理,处理的结果给研究人员提供了一个客观可靠的数据基础,这大大地减少人工检阅和人工处理等繁杂的工作,提高了研究者们的效率,同时这些数据基础上研究人员们能够更深一步的论证、假设、探索、实验。从而加快了生物医学领域的发展进程。
第二章 入门概念
2.1 蛋白质以及翻译后修饰概述
蛋白质(protein)是生命的物质基础,是有机大分子,是构成细胞的基本有机物,是生命活动的主要承担者。中学课本学习过,蛋白质是由氨基酸构成的,而氨基酸是由基因表达产生的。所以可以认为蛋白质是基因表达的产物。蛋白质翻译是蛋白质合成过程中的第一步,翻译是根据遗传密码的中心法则,将成熟的信使RNA分子(由DNA通过转录而生成)中“碱基的排列顺序”(核苷酸序列)解码,并生成对应的特定氨基酸序列的过程。此过程产生的蛋白质是没有活性的,想要具有活性,常常要进行一个系列的翻译后修饰和加工,从而使其具有一定的功能和特性。据研究统计,人类有50%~90%的蛋白质都在翻译后进行了修饰[6]。蛋白质的翻译后的化学修饰一般是对蛋白质的附加一些生物化学官能团或是改变其结构来让它改变其化学性质,从而让它具有某项特性或功能。翻译后的蛋白质正因为有了修饰加工这一个步骤,就可能使相同的蛋白质具有完全不一样的功能和特性,也大大地增加的蛋白质的复杂性和多样性。所以这种类远远地超过了有天然20中氨基酸经过排列组合可生成的蛋白质的种类。所以,一个基因表达产生一个蛋白的说法就不存在了。据统计,人体类的可以编码合成蛋白质的基因组有两万多个,但是却可以表达出高达20万种不同功能的蛋白质[6]。
到目前已经发现了超过400种的蛋白质翻译后修饰的方式。但是目前发现的蛋白质翻译后修饰的方式还是比较少的,还有大量的蛋白质修饰过程还是未知的。然而我们比较常见的修饰方式有磷酸化、甲基化、泛素化、乙酰化、亚硝酸化、糖基化、氧化等[7]。
2.2文本挖掘技术概述
文本挖掘(Text Mining)是以计算语言学、统计数理分析为理论基础 ,结合机器学习和信息检 索技术 ,从文本数据中发现和提取独立于用户信息需求的文档集中的隐含知识。它是一个从文本信息描述到选取提取模式 ,最终形成用户可理解的信息知识的过程[8]。它其实也是数据挖掘技术的一个分支学科,它和数据挖掘不同的是,数据挖掘技术主要是对结构化的数据进行挖掘处理,比如说关系表(Table),事务表等,这些表里面的数据(特征)在积累的时候就已经通过键值对(key-map)的方式做好了结构化的存储,所以计算机在解析数据的时候根据不同key去取相应的数据,所以结构化的数据是能被计算机所识别的,但是文本挖掘技术主要针对的是文本形式存储的非结构化数据,比如说新闻,网页,书籍等,这些文本数据是通常是由人类所撰写,文本中充满了人类语言规则和定义,这些规则因地区而异,比如说不同国家的语言不一样,计算机必须为每种语言去适应其语法规则,或者时间而异,比如说随着科研不断发展,新的名词可能被创造,这个时候必须让计算机学习人类创造的新的词语,由此可见计算机是无法直接识别这些非结构化的数据。
2.3 Pubmed生物医学文献检索系统
Pubmed是美国国家医学图书馆(NLM)所属的国家生物技术信息中心(NCBI)于2000年4月开发的,基于WEB的生物医学信息检索系统。PubMed的数据库来源于MEDLINE,OLDMEDLINE,Publisher-Supplied Citations,其中包含了大量的生物医学先关的期刊,论文,综述等[9]。它给生物医学研究人员提供了丰富的有关蛋白质,基因等重要的文献信息。
第三章 需求分析和系统设计
3.1 需求
需求最初是由中山大学生物工程学院的几位师姐提出来的,她们研究蛋白质翻译后修饰的时候会经常去Pubmed生物医学文献检索系统里面去找有关蛋白质翻译后的文献,比如想要找找有关蛋白质磷酸化修饰的文献,学习这一类修饰后的蛋白质的知识,她们可以在Pubmed文献检索系统里输入phosphorylation关键字,接着就可以找到几千或者上万篇有关磷酸化的一些文献。这些根据关键字检索到的文献的数量有点庞大,人工每一篇地去阅读显得有些吃力,但是她们发现她们每次阅读文献的时候都有一个共同点,就是只要找出文献里面的关键信息,比如说只要找出某某蛋白质被某某酶在某个位点上修饰了。这样问题就来了,能不能借助计算机来识别这些关键信息并找出这些关键信息呢?如果可以的话就不用通篇阅读了,大大地节省了时间,提高了效率。于是她们就提出了这样一个需求:通过计算机来识别并找出文献中的一些关键信息,关键信息就是蛋白质的名称,酶的名称,酶修饰蛋白质的位点。
3.2 需求分析
接到这个需求的时候,给我的第一感觉就是要在一篇文章中找出摘要信息,用几个关键词概括出全文,这有点类似于文章目录提取和文章摘要概括。于是我去Pubmed数据库查了几篇有关于磷酸化修饰的文章。文章的主要的意思就是描述了某个激酶在某个位点上修饰了某个蛋白质,当然也有一些是无关紧要的文章,这些无关紧要的文章我们要把它们除去,因为它们并不能给我们带来知识和信息。图3-1是一篇标准的Pubmed数据库中关于磷酸化修饰的一篇文章。在示例文中以标签ArticleTitle为例,ArticleTitle内容为GRK6 phosphorylates IκBα at Ser(32)/Ser(36) and enhances TNF-α-induced inflammation.在这句话中GRK6是一种激酶,IκBα是一种蛋白质,Ser(32)/Ser(36)是修饰的位点,它们三者之间的关系是GRK6激酶在Ser(32)/Ser(36)位点上磷酸化修饰了IκBα蛋白质。这就是研究人员想要找出的知识模型。

3.3 用户用例分析
需求我们已经知晓了,在这基础上,我们要建立一条完整地用户使用示例,这样我们才能了解整个系统的来龙去脉。假设一个用户需要在Pubmed生物医学文献检索系统中找出蛋白质磷酸化修饰的一些信息,用户可能想要输入关键词phosphorylate(磷酸化),然后用户想得到所有(当然不可能找出所有的被磷酸化修饰的蛋白质,有些蛋白质研究人员们还在进一步在探索,这里的所有指的是目前已经发现的并且已经被收录于MEDLINE文献数据库中的所有被磷酸化修饰过的蛋白质)被磷酸化修饰过的蛋白质,以及修饰的激酶和修饰的位点,并且这些数据以一种直观、可视化的图表来展示在他面前。简单抽象一点概括的话,用户输入一个pubmed文献,然后期望得到一个可视化的图表。图3-2展示了用户和系统这样一种关系。

根据用户用例分析,接来下把用户和系统交互的流程按照时间顺序来列出来,于是可以设计出时序图如图3-3所示。

3.4 系统设计
3.4.1 系统工作流程和场景
根据以上需求描述和用户用例分析,我们首先可以建立起用户和系统的工作场景,图3-4展示了用户和整个系统的一个工作场景。从图中可以看出整个工作环境下有两大类,一个是客户,一个是服务,这就是典型的客户端-服务端(C/S)架构的模式,在论文中稍后会提及到这种架构。从图发现整个系统主要处理工作流程的是在服务端,于是结合上述所有,我把此次的系统工作流程大致的分成以下5个步骤:
-
从Pubmed文献数据库中获取有关蛋白质磷酸化修饰相关的文献
-
对这些原始文献文本做一次预处理,整理出具有一定格式的文档集
-
针对于文档集中的每一个文档,识别出文档里面的命名实体
-
挖掘出这些命名实体之间的关系
-
对挖掘出来的数据和关系进行可视化处理

3.4.2 模块化设计
根据以上总结的5大工作流程,我们可以发现以上5个步骤顺序执行,下一个步骤总是依赖上一个步骤,并使用上一个步骤产生的数据作为这一步的输入数据。每一个步骤都有一个输入数据和输出数据,由上往下以此传递。所以我们在实现整套系统的时候要依次实现这5个步骤,并且每个步骤都有一个input和output。每一个步骤都有一个明确的功能,我们可以把每一个步骤对应地划分成系统的各个模块,然后以其功能来命名。这样我们就可以把它划分成文本预处理模块,命名实体识别模块,实体关系提取模块,数据可视化模块五大模块。系统整体设计图如图3-6所示。由图3-5可以看出各个模块的输入和输出总结如下表格图3-5所示。
| 模块名称 | 输入数据 | 输出数据 |
|---|---|---|
| 文本预处理 | 粗糙、冗余的文件 | 具有一定格式的文档集 |
| 命名实体识别 | 具有一定格式的文档集 | 被标注命名实体的文档集 |
| 实体关系提取 | 被标注命名实体的文档集 | 实体、关系 |
| 数据可视化 | 实体,关系 | 实体+关系图表(统计) |

各个模块内部遵循模块相互独立原则,仅在模块的出口和入口提供对外数据交接的接口,这样就便于以后维护方便。
3.4.3 软件开发架构
从系统的使用场景图3-4上来看,我们采用C/S(客户端/服务器)的架构来实现整套系统,因为从用户用例上,这是基于请求—响应式的模型的事件过程,这也是典型的C/S架构的模型,所以采取C/S架构是非常贴切的。
由于文本集数量庞大,在处理这么多文档集的时候会非常耗时,所以显然不能时时同步响应用户的请求,当然也不能处理完所有的文档再响应用户的请求,因为处理完所有的文档集再返回给用户,用户就回觉得系统响应非常慢,非常卡,从而造成用户体验效果不佳。所以我们服务端方面要每处理完一篇文档的时候就及时的返回数据给用户,直至处理完所有的文档集。这样的话,有数据时时返回,虽然不是全部的数据,但是用户就不会觉得卡,反应慢。客户端方面,要做到异步加载服务端传回的数据并展示。
为了加快系统的运行效率,服务端在处理文档集的时候应当采用多线程的方式,并发处理每一篇文献,因为在处理每一篇文献的事件是相互独立的,前提是每个线程在处理资源的时候,不能同时访问同一个文件,因为这样的话,可能会导致结果有重复,所以在线程调度方面,对于同一个文件的处理的时候要加上互斥锁,以保证每个线程在相同时刻都在处理不同的文件。
3.4.4 开发环境
-
开发语言;Java
-
操作系统:MacOS
Java是一种跨平台式的语言,可以方便移植到不同平台使用,所以此次开发主要采用Java语言。采用J2EE里的JSP和java servlet作为服务端的开发架构,客户端就可以认为是H5网页前端。
第四章 程序设计和系统实现
4.1 程序设计
程序的主体是服务端,而客户端简单的来说就是前端H5网页通过浏览器就可以打开,所以服务端最后返回的数据是html,css,js等等。服务端的开发采用的是Jsp+Tomcat技术。图4-1展示了服务端内部的一个大致的结构,从图中可以看出来整个程序分文三个大区域,第一是资源区,这个主要是托管一些文档、缓存、数据库这样一些体积比较庞大的资源,第二个是主程序逻辑控制区,这个区域是运行的是程序的主线的逻辑,从请求的开始到返回这里面复杂的逻辑过程都是由主线来严格控制。第三个区是文档处理区,由于文档处理这一步非常耗时,里面的逻辑也非常复杂,但是功能相对独立,所以我就把它单独列出来,相当于是程序的一个分支。

4.2 文本数据源获取
由于需求提出者提出了主要针对蛋白质磷酸化修饰的信息挖掘,所以关键词即为’磷酸化’(phosphorylate)。Pubmed文献检索系统上收录的大部分是以英文为语言的生物医学文献,所以我们要把关键字’磷酸化’翻译成phosphorylate来作为Pubmed文献检索系统的关键词。Pubmed文献检索系统支持AND和OR来接连关键词,这样就可以限制和放宽检索的范围,因为此次研究的对象是整个和磷酸化有关的蛋白质文献,所以我们在输入关键词的时候把磷酸化的主动式(phosphorylate)和被动式(phosphorylated)两个词通过OR的方式来扩大检索的范围,通过检索后,最后我们从中得到了76604个文献文本结果。文献结果以XML的文件格式下载到本地文件系统上面,这样我们就得到了一个最原始的文本数据。
通过以上方法得到了单一的文件,里面包含了76604篇有关磷酸化的文献,文件大小1.05GB,一共2000多万行,这么大的文件肯定不能同时加载到内存里去处理,所以我就把这么大的文件拆分成20个50多M的小文件,这样每个文件里包含了3000-4000篇的文献,用1-20来对每个拆分过的文件命名,这样下来,我们就得到了20个XML文件如图4-3所示,这20个文件就是我们文本数据源,也是文本数据获取模块的输出,文本预处理模块的输入。

4.3 文本预处理
文本预处理是文本挖掘中的一个关键的步骤,预处理的结果可以直接影响到最终挖出数据的质量和效果,所以这一步非常关键。文本预处理的主要的思想就是把源文本数据简单化,结构化,便于下一步的处理。
经过上一步的处理,已经得到了源文本数据,这一步就对这些源文本数据作进一步的处理。仔细观察这20个XML文件的内容,图4-3展示了一篇XML的大致格式,发现里面对一篇文献描述的时候附加了很多的信息,比如说创建日期、收录日期、公司版权、作者等等,这些信息都是我们不关注的,无关紧要的信息,但是却占用非常大的空间内存,同时这些冗余的数据可能还会对接下来的文本处理带来不必要的麻烦。我们只关注数据是这篇文献的标题,文献内容,pubmed_ID标示符,所以在文本预处理的阶段,我们要把这些冗余,无关紧要的数据给去掉,只要求保留Pubmed标示符、文献标题、文献内容这三个关键数据。这样下来,我们就做到简化了数据的目的。接下来我们要使这些文本数据具有一定的结构(这里的结构化并不是只能被计算机直接识别的那种结构化)。于是我们把每一个文件里面的3000-4000文献拆分成3000-4000个小txt文档,txt文档的文件名就是序号加文献标题,txt文档里的内容就是文献内容,于是我们就可以得到70000多个文献文档,这就是我们对文本预处理之后得到的文档集(Document Set),每一篇文档都对应于一篇文献,并且包含了文献的标题和内容。

用Java程序实现上述的过程。首先XML文件的内容是一种它本身就是一个标示性可扩展标记语言(Extensible Markup Language),我们可以通过一些java开源库提供的API来对其进行解析。dom4j是一个非常优秀的JAVA开源库,处理的效率高,对于这么庞大的文本数据来说,dom4j的处理比其他的开源库的效果会更好。首先对20个XML文件进行for遍历,对于每一个文件,遍历每一篇文献从中解析得到一些属性和标签,由于只要文献标题和文献内容,所以直接取和这两个标签下面的内容,然后再把内容输出到新建文档中,文档命名为序号。如果我们只在单线程拆分这些文件的话,效率可能不够高,我们也可以用多线程来预处理,用多线程的话,就不用for遍历这20个文件了,可以直接启动20个线程,每个线程里处理一个文件,启动20个线程的好处就是每个线程都处理不同的文件,不会发生资源互斥的问题。
代码里面我先创建一个XMLParser静态工具类,里面全部实现两个静态方法,一个是解析XML得到里面的文献标题articleTitle和文献内容articleContent,另个是把文献标题和内容输出到一个文本文档里面,构成文档集中的一个文档。整个预处理过程的伪代码如图4-4所示。整个处理过程代码中有四个值得注意的地方,一是XML文件里面有些文献的标题的长度非常长,超过了linux文件系统下面支持的最大文件名称长度256字节,所以如果出现超过256的字节要进行相应的裁剪。二是有些PubmedArticle标签下没有包含Abstract标签,所以在处理的时候要加以判断,如果Abstract为空的话,那么就跳过这篇文献的处理,如果还继续输出一个空白的txt文档的话,空的文档对我们的数据挖掘根本没有贡献,反而在文本挖掘的时候或者统计的时候还可能会导致误差。三是有些Abstract标签下面会包含多个子标签,这些子标签里面的内容都是文献的内容,所以在输出的时候要把他们全部输出,不仅仅只输出单个的AbstractText。四是文献标题字符串中会有一些”/”这样的字符,我们把它作为文件名称输出的话,java的File类会认为这是一个子目录的标示符(类似dir/sub里面’/’表示的子目录的意思),所以在处理文献标题的时候要把标题里面的’/’字符用:代替,这样File类就会把这当成文件名称去处理。
//文档预处理-生成文档集
main(){
//第一步解析xml文件,把每一篇文献存入List中
List pubmedArticleList = parsexml(xmlFile);
//第二步,对于xml里面的每一篇,都要把它写入文档及文件夹下面
for each pubmedArticle in pubmedArticleList{
addToDocSet(pubmedArticle,docSetPath);
}
}
//对于每一篇article增加到文档集
addToDocSe(pubmedArticle,docSetPath){
//1.首先要指定一下文档的名字
//这里采用pmid + title的形式来作为文件名
fileName = pubmedArticle.pmid + pubmedArticle.title;
//2.建立文本文档
//文档的内容为pubmedArticle.content
createFile(fileName,pubmedArticle.content,docSetPath);
}
4.4 命名实体识别和实体关系提取
4.4.1 命名实体识别概述
命名实体识别是文本挖掘过程中一个重要的过程,它在文本挖掘中的作用就是找出文本中具有特定意义的实体词语,比如说人名,歌名,影视名等等,同样地,生物医学命名实体识别(Biomedical Named Entity Recognition, Biomedical NER)也是生物医学文本挖掘过程中的关键任务,其目的是从生物医学文本集合中识别出指定类型的名称, 如蛋白质、基因、核糖核酸、脱氧核糖核酸等[10]。这是进一步抽取关系和其他潜在信息的关键步骤。
命名实体识别是整个文本挖掘过程中的关键任务,因为有了识别出的实体,接下来的步骤才可能得到执行,同时它也是非常复杂的任务,里面夹杂着概率统计学,人工智能,机器学习,自然语言处理等各方面的学科知识。目前命名实体识别的方法主要分成三类,一是基于规则的方法,二是基于词典的方法,三是基于概率统计的方法[11],基于规则的方法就是特定的实体词语在文本中总是以一定的格式和规则呈现的,比如说书名,它一般是出现在《》书名号里面的。大多数的实体名词都是没有明显的特征和规则的,所以这种基于规则的实体命名方法非常不精准,而且局限性很大,但是它简单快捷。第二种命名实体识别的方法是基于词典的,它的思想就是把所有我们想研究的实体名词都收录在一本词典上,当在解析文本的时候就回去词典库里面去进行匹配,就像我们去查字典一样,这一方法需要人工录入巨量的词语到词典库里,它在识别词语的时候不会根据上下文的语境来识别,更加不会识别一词多义,有歧义的语法情况,只能匹配词典库里的词语,词典库里面有就能匹配,没有就不能匹配,所以这种方法在处理人类语言的时候不免捉襟见肘,漏洞百出。第三种实体识别的方法是基于概率统计的方法,也可以说成是基于机器学习的方法,这种方法也是在当今命名实体识别领域里面研究的重点,因为这种方法能够较为精确地识别出文本中的实体名词。它能够根据上下文语义环境来识别出里面的文本中的实体,并且能够识别出一词多义的词语。它的主要思想就是我们人工的将训练文档集中的实体名词找出来,并给其赋予一个词性标注,再根据上下文关系统计出一套可以让计算机学习的识别模型(语料库),然后计算机就能够根据这个模型来识别出未知文本中的实体名词了[12]。这样说的比较抽象,打个比方,我们要识别south china university of technology(华南理工大学)这个学校实体名词,首先我们的(识别模型)语料库里面有了这个词语,并且这个词语我们已经给与了其学校词性的标注,统计出了五个单词中每个单词出现在这五个单词中的概率和出现在整个语料库的次数,接下来有了这套概率,计算机在识别未知文本中的south | china | university | of | technology词语的时候就每个单词出现在上下文的环境下的概率最高的就是当它出现在语料库中词组south china university of technology学校实体名词,这样计算机就识别出了一个学校实体名词。目前有很多种建立语料库的概率模型,如贝叶斯模型、隐马尔可夫模型、支持向量机等等[13],这些方法都需要大量的训练数据,但是相对准确。
4.4.2 ABNER命名实体识别工具
生物医学领域上的应用非常广泛,ABNER,BallE等工具是比较出著名的命名实体识别的工具[11],本文以ABNER为实验工具来进行实验和开发,我们选择它的原因主要是第一,它是基于JAVA平台下开发的软件;第二,它是一个开源的系统;第三,它能够提供给我们JAVA的API接口,可以让我们进行第二次开发,我们能够把它的代码嵌入到我们的系统中去。图4-5是Abner工具一个简单的界面。

ABNER是一款用于生物医学文本分析的软件,主要用于识别生物医学实体,它所使用的识别的方法就是基于统计概率的方法,首先它包含了NLPBA和BioCreative两个庞大的语料库其中包含了绝大多数已经发现的生物医学的实体,这两个语料库的F1测度分别是70.5和69.9[14]。它目前能够识别出得实体的类型大致有五类:蛋白质,DNA,RNA, 细胞系,细胞类型。此外,它不仅提供我们识别命名实体的接口,还提供了训练数据集、培养语料库的接口,所以理论来说,我们只要有足够多的语料库,我们就可以识别更多类型的实体,也不仅仅仅限于生物医学实体的识别。ABNER本质上就是一个基于概率统计的机器学习系统,采用了基于上下文特征的线性条件随机场的方法,线性条件随机场方法又是建立在隐马尔科夫模型(Hidden Markov Model,HMM)上的[15],隐马尔科夫模型是一个可以通过可观察到的状态(好比文本单词,这是我们可以观察到的状态)根据相关概率去发现隐藏的状态(好比文本单词的词性标注,单词词性标注是我们未知),所以如果发现了未知文本的词性标注,就可以识别出了实体。
应用ANBER工具到我们的系统。首先我们进入ANBER网站ABNER: A Biomedical Named Entity Recognizer [16],下载abner.jar可执行的jar包,这也是可以依赖的三方库,图4-6展示了这个jar包里面重要的类的说明。如果直接运行的话,可以得到如图4-5的画面,输入一段待识别的文本,按下annotation按钮,它就可以识别出了文本中的生物医学实体,并且用不同的颜色进行标注和高亮显示出来。
| Input2TokenSequence | Input2TokenSequence is a text processing Pipe for the MALLET framework. |
|---|---|
| Scanner | ABNER's Scanner class implements the finite state machine used in tokenization. |
| Tagger | This is the interface to the CRF that does named entity tagging. |
| Trainer | The Trainer class will train a CRF to extract entities from a customized dataset. |
现在我把它通过三方库的方式引用到我的工程里面。首先实例化一个Tagger的对象Tagger t = new Tagger();再条用Tagger里面的方法getEntities(fileContent, "PROTEIN");函数里面的第一个参数表示文献文本字符串,第二个是具体的哪一个标签,我们要识别文本里面的蛋白质实体就可以用"PROTEIN"作为函数的第二个参数。这样我们就可以获取文献里面所有的蛋白质的实体,图4-7是部分关键的代码:
//实例化tagger对象
Tagger t = new Tagger();
//调用API方法,获得文中的实体
String protein[] = t.getEntities(fileContent, "PROTEIN");
由于系统还要挖掘出与蛋白质磷酸化修饰有关的激酶和修饰位点,所以我们必须还要继续识别文本中的激酶和修饰位点的实体名词,但是ABNER工具只提供了蛋白质,DNA,RNA, 细胞系,细胞类型五个类型的实体,所以我们还要对这些实体进行训练,培养一个能够识别激酶和修饰位点的语料库。ABNER工具给我们提供了训练语料库的接口。我们可以初始化一个Trainer的实例,调用Train里面的train(trainFile, modelFile)方法,trainFile就是训练文本的路径,modelFile就是语料库模型文件的路径,比如想识别一个磷酸化修饰的位点(Ser32),我们可以在trainFile的文件里面填写训练的规则,官方的demo里面介绍了如何填写训练集文件,图4-8是官网demo的一个训练格式。训练文件的规则概括成四点:一,每一句话要按照每个token(可以理解为每个单词)空格间开,并且一句话写在一行;二,每一个token的左边的单词,右边是词性标注,两者用‘|’符号间隔开;三,每一个实体的第一个单词的词性标注前要加前缀B-,剩下其他的单词前面要加前缀I-;四,每个不是实体的单词的词性标注要设为O。用着我们训练一段能够识别位点的训练集如图4-9所示。生成一个model,然后用model作为语料库去识别出文档里面的为位点实体。图4-10中的代码说明了如何训练一个语料库,并使用语料库进行识别文档的。
<pre>
IL-2|B-DNA gene|I-DNA expression|O and|O NF-kappa|B-PROTEIN B|I-PROTEIN activation|O ...
</pre>
GRK6|O phosphorylates|O IκBα|O at|O Ser32|B-POSITION or|O Ser36|B-POSITION and|O enhances|O TNF-α-induced|O inflammation|O .|O
//第一步 训练得到语料库
Trainer t = new Trainer();
t.train(trainTextPath, modelTextPath);
//第二步,用训练出来的语料库去识别文本
Tagger t = new Tagger(new File(Config.MODEL_PATH));
String res[] = t.getEntities("GRK6 phosphorylates at Ser32 and enhances TNF-α-induced inflammation.", "POSITION");
经过以上的步骤,我们可以通过abner里面自带的语料库去识别文本中的命名实体,也可以自己训练想要识别的实体的语料库来识别。
4.5 实体关系提取
4.5.1 关系提取概念
经过命名实体识别之后,现在就要提取实体之间的关系,这一步称之为实体关系提取。实体关系的提取性能的关键因素在于实体识别的准确率,实体识别越准确,关系提取的性能就越高。目前主要有五种实体关系提取的方法[17],第一种是基于模式识别的提取方法,基于模式识别的方法是普遍使用的提取方法,这种方法主要使用了语言学的知识,在提取实体关系之前,先预先定义出一些基于句法、语义、等一些结构的模式,然后在提取的过程中,把待处理的文本片段与这些模式进行匹配,然后通过一定的计算得出是否匹配,如果匹配则这文本片段具有该模式所包含的关系特征,从而可以判断出改文本中的实体之间具有某种那个关系;第二种是基于词典驱动的提取方法,基于词典的方法相比模式匹配的方法具有较高的复用性,用户想匹配新的实体关系的时候不用具备所有的语法,语义模式,而只需往词典里面添加新的关系词语,系统即可根据词典来提取相应的关系;第三种是基于机器学习的提取方法,这种方法的核心思想就是把实体关系提取看做是一种分类,进而关系提取就相当于是一次类别的判断。运用此方法之前我们需要手动地对分类器培养一批语料库,然后分类器应用这些语料库对文本进行分类,从而得出相应的关系;第四种基于Ontology的 提取方法,最后一种是混合式的提取方法。
4.5.2 Rlims-p工具介绍及其工作原理
Rlims-p是一款典型的基于规则的文本挖掘工具[18],它是针对于挖掘提取蛋白质磷酸化修饰的信息,包括蛋白质底物信息,修饰激酶信息,修饰位点信息,还包括了前三者之间的关系信息等等。本篇论文中的核心文本处理的机制就是围绕着Rlims-p程序展开的。采取此工具的原因主要概况为两点,一是从功能上来说,这款工具恰好完全能够帮助我提取文献里的实体,并且找出其中的关系,这也是本次论文设计的主要目的,二是这款工具给我们开发者提供了Rlims-p文本处理的接口,只要把文献的内容通过传参的形式发送至对方的服务器,对方就会及时处理并且把结果返回,这样就可以在rlims-p的帮助下进行二次开发。
Rlims-p工具里面主要包含两个主要的模块构成,第一个模块是自然语言处理模块(NLP Pipeline),第二个模块是信息提取模块。每个模块里面有分很多个处理步骤。图4-11展示了Rlims-p的模块结构图[19]。

第一个自然语言处理模块其实就类似于一个命名实体识别的一个过程,输入一个文档集进去,输出的结果就是文本里每个短语的词性,当然这个短语并不一定就是命名实体,词性就是描述这个短语在上下文中的类型,比如说是动词,名词,基因,蛋白质,蛋白质附属物,化学物质等等。自然语言处理模块内部又分为了若干的小步骤包括了格式化数据,句子拆分,token化,单词标注,分词,短语类型标注等等,这些标注对第二个模块里面的模式匹配有着重要的作用,稍后再提到。第二个信息提取模块就是把第一个模块里的输出作为输入,然后提取出文中短语之间的关系,判断出短语具体是蛋白质还是激酶还是位点等等,最后输入的就是检索的标注了,这个标注里面包含了,各个蛋白质,激酶,位点的名称,以及实体之间的关系。这个模块所使用到提取关系的方式就是基于规则的关系提取方法。首先它预先定义了触发器-参数模式,扩展模式,链接关系模式三大模式,然后根据这三大模式再在文本中进行模式匹配,根据匹配的结果再进行判断。
模式一:触发器-参数模式,它是最普遍的模式,里面包含了名词加动词的语法结构,比如说A phosphorylate B,A phosphorylate B in C,B phosphorylated by A in C等等,phosphorylate是触发器,A,B,C都是参数。根据语法规则,在描述蛋白质磷酸化的句子中语法结构一般都是激酶磷酸化了蛋白质在某某位点上,所以在上述我们可以大概推出A可能是激酶,然后再结合A的类型(第一个模块里面已经获取了短语A的词语类型)加以判断,如果A类型是蛋白质类型,那么就认为A就是一种激酶(激酶也是一种特殊的蛋白质)。在语法结构A phosphorylate B中,根据英语的语法我们可以大概退出B可能是被修饰的蛋白质,也可能是位点,那B具体是啥,这样根据模块一种得出的短语B的词语类型,假如说B是蛋白质类型,那么B就是被修饰的蛋白质,如果B是蛋白质上的附属物,那么B就是位点(位点是蛋白质上的一部分)。由此可见在这个模块里不仅可以发现短语之间的关系,还能够确定短语究竟是属于被修饰的蛋白质,还是激酶,还是位点。模式二:扩展模式,由于英文语法的多样性,表示这三者之间的关系不仅仅只靠以上几种语法结构就能匹配的,于是又新增了很多扩展的模式来匹配,模式三:链接关系模式,在英文语法中会出现这样的情况,A phosphorylate protein such as x,xxx,protein先行再前,其真正的主体是x,xxx,所以protein和x,xxx有着链接的关系。这样的先行词和先行词后面紧跟定义的链接关系还有很多种,所以在这里必须要考虑这样一种情况。
4.5.3 嵌入使用Rlims-p工具
打开Rlims-p工具的网址RLIMS-P [PIR - Protein Information Resource] ,点击页面的Web Service超链接,他给我们提供了一个网页服务编程接口,我们可以通过Http请求中的post方法并把待处理的文本数据转换成post参数,发送至服务地址http://annotation.dbi.udel.edu/text_mining/bioc/bioc.php 上[20],服务器就回处理并返回一个XML格式的文本文件,里面包含了处理之后得到的实体名称,和实体之间的关系。
4.6 多线程处理文档优化
考虑到我们文档集中的文件数量非常庞大,如果在同一个线程里面处理这么庞大的文件的话将会非常耗时,所以我不得不采用多线程的方式来并发的处理文档集里面所有的文档。JAVA语言特性也是支持多线程开发的,更方便的是JAVA库java.util.concurrent中提供了线程池的工具,方便我们管理和调度线程。为了避免多个线程同时处理同一个文档,尽量让每个线程都独立,互斥的运行,我设计了两套解决方案,第一种方案是在同一时候只允许一个线程去文档集里面取文档,并把文件标记为已读,取到要处理的文档之后各个线程该干嘛就干嘛,互不影响,直到文档集全部处理完毕。(PS : 取文档只是一个获取对文档的引用标识,耗时远远低于处理文档的时候,所以同一时刻只有一个线程获取文档的引用并不影响程序的效率)。Java运用synchronized关键字和同步锁的机制可以实现同一时刻只用一个线程在取文件,线程在执行带有关键字synchronized的方法的时候会检查一下同步锁有没有被释放,没有释放就表示正有线程在运行这个方法,就会一直等待直到同步锁被释放再运行。第二种方案是首先预先分配线程池的大小,然后再把文档分给各个线程去处理,每个线程要处理的文件的数量都保持差不多的水平。比如说,我有一万个文档要处理,文档编号从1-10000,那么首先分配一个最大支持50个线程同时运行的线程池,再给每个线程分配200个待处理文档,文档的序号要分段开来,线程1处理1-200的文档,线程2处理201-400的文档,依次类推,直到所有的文档集都分配完成,这样一来每个线程都知道自己要处理哪一批文档,不会发生越界的情况。但是第二种方法的不好之处在于假如某个线程所处理的文档内容非常小,一下子就处理完毕了,那么这个时候这么线程就结束了,它不会去为别的线程分担压力了,线程池里面就少了一个线程,就不能时时刻刻发挥线程池处理的最大能力了,而第一种方案,线程池里面时时刻刻都能够保持最大的线程数量,除非待处理的文档数量少于线程数量了。形象地来说,第一种方案像一个非常团结的团队,团队中的每一个人做完了一个任务后就争着去抢下一个任务做,直到任务全部完成为止。第二种方案像一个自私的团队,团队中的每一个人随机的领了相同数量的任务,任务做得快的人就早早地下班了,不会帮别人分担,任务做得慢的人就加班加点完成任务,但是最终的效率是由最后一个完成任务的人决定的,所以从效率来说显然是第一种好过第二种,所以在程序设计的时候,我采用的是第一种方案,图4-12展示了方案一的处理流程图。Pubmed文献库中和磷酸化先关的文献有20多万篇,如果只用一个线程去处理的话,再加上用Rlims-p工具本身就非常耗时的网络请求的话,那么处理这20万篇的文献的耗时将会达几十天,如果处理的文献数量达到几百几千万篇的时候,那么我相信一台电脑的计算能力就不太够用了,可能就要用到分布式机制的才能够提高处理的效率。

4.7 文档预处理和缓存机制

在客户端发起的请求,服务端要处理一段时间后才会返回给用户,这个时候用户就需要等待了,文档数量如此庞大,如果有很多个用户同时来请求的话,那服务器处理的时间就会变得很长,返回给用户的时间也就会变得很长,为了解决此问题,提升程序的用户体验,我们要预先对所有的文档进行处理,并且把处理后文档的结果保存再缓存区里面,然后每次用户请求的时候,服务端这边直接把缓存区里面的结果直接返回给用户,这样的话就减少了很多用户等待的时间,图4-13展示了缓存机制的一个逻辑流程图。第一步我们需要对文档进行预处理,预先找出文档里面的实体和关系,这一步也需要用到先前提到的多线程的处理方式来处理,然后把这些结果通过文件的形式保存在文件系统里面,通过文件名来作其的键值key,文件内容则是value。在JAVA里面把一个java Object(文档处理结果)输出成一个文件,用到了序列化的机制,通过让这个java object实现序列化,然后通过FileOutPut把它以字节流的方式输出,最后得到的文件就是我们的缓存文件。有了缓存之后,用户请求发送过来之后,首先我们会根据用户发送过来想要处理的文档的id来去缓存文件夹下面去找对应的缓存文件,如果找到了就把这个结果返回给用户,如果没找到的话就按照正常的流程来处理文件,处理完成之后再把处理结果存进缓存文件夹,这样一来,对于文档集新增的文档还没来得及做一次预处理,这个时候用户只需再第一次请求的时候进行等待,之后的请求都可以从缓存直接获取不用等待了。
第五章 数据库设计和数据可视化
5.1 数据库设计
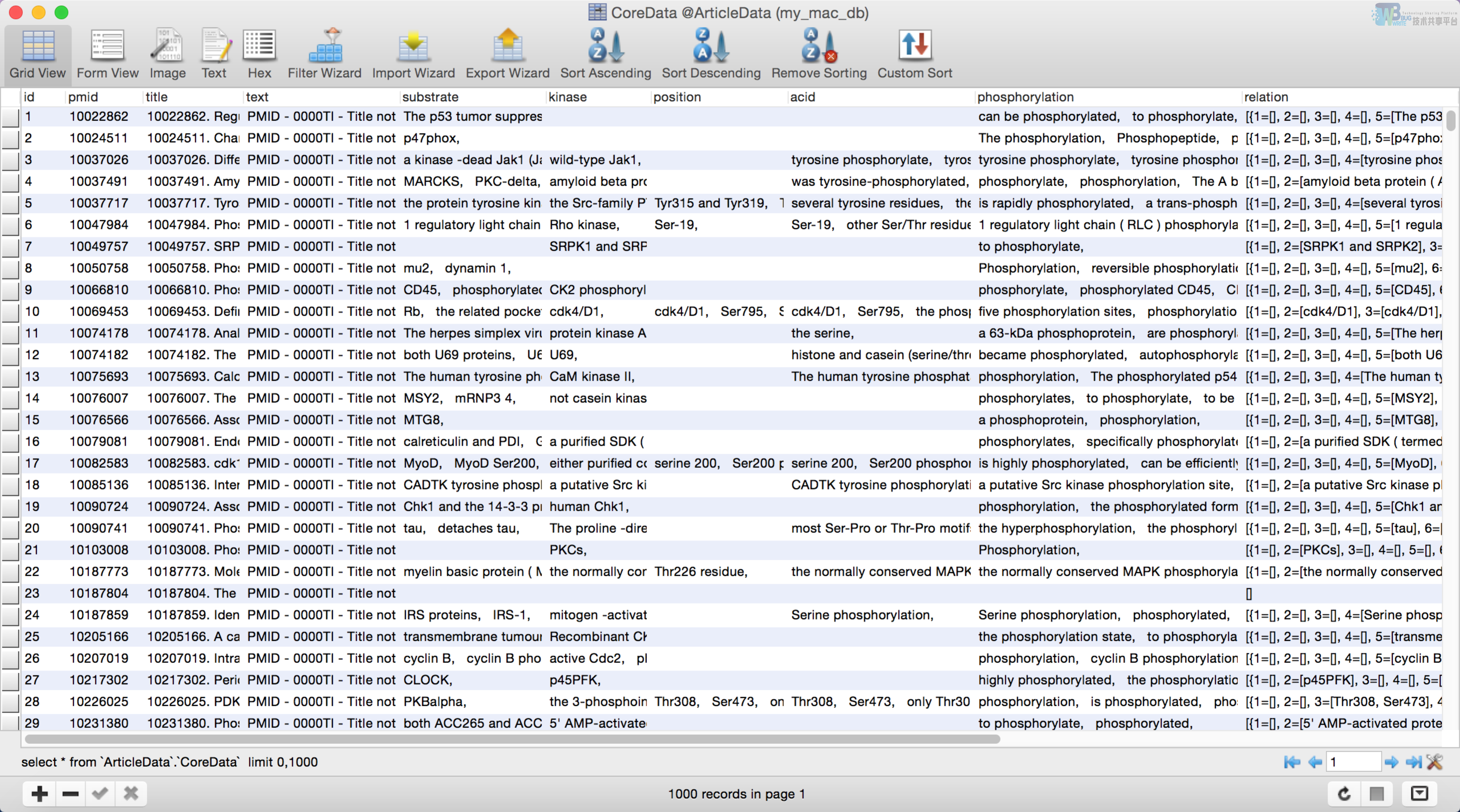
从需求上来看,需求是要从给定的pubmed文献中挖掘出文本中的被修饰的蛋白质,激酶,和位点这三种实体,以及这三种实体之间的关系。由此我们可以归纳出数据库中表里的字段,表5-1给出了表中的字段以及字段的含义。
| Id | 自动增长的id,主键 |
|---|---|
| Pmid | pubmed文献在文献检索系统中的id |
| Title | pubmed文献的标题 |
| Text | Pubmed文献的内容 |
| Substrate | 被修饰的蛋白质 |
| Kinase | 修饰蛋白质的激酶 |
| Position | 修饰蛋白质的位点 |
| Acid | 文中氨基酸实体 |
| phosphorylation | 磷酸化的词语或短语 |
| Relation | 实体之间的磷酸化的关系 |
表中一共有十个字段,其中蛋白质,激酶,位点等这种实体的每个字段可能有多个实体短语,所以我们规定这些短语之间用逗号隔开,另外同一个实体可能出现在不同的字段里面,例如实体A在关系R1里面充当了蛋白质的角色,而在关系R2里面充当了激酶的角色(激酶也是一种特殊的蛋白质),这样的话实体A既出现在蛋白质字段里面,也出现在激酶字段里面。实体关系这个字段比较特殊,因为文献中可能包含多种关系,或者不包含关系,而每一个关系包含了五种类型的实体,为了简单起见我们还是把这个关系保存在一个字段里面,而不是新建一个表专门用来存储关系,首先relation字段在程序里面的数据结构是一个List,List中的每一个element就是每一种不同的关系,关系element的数据结构又是一个map<key,value>,key就是字段名,value就是识别出来的实体。综合来说关系在程序中的数据结构就是List,在java程序中采用List中的toString成员方法可以把关系结构转换成一个字符串,再把这个字符串保存至数据库。
本系统采用的是mysql数据库,通过jdbc驱动实现了java代码新建数据库,表,插入数据等操作。往数据库插入数据的时机和缓存是一样的,首先是预缓存一遍,同一样也是预插入数据库一遍,然后对于文档集中新增的文档,如果没缓存的话,用户在请求处理此新增的文档后,服务端就会对新增文档处理的结果进行一次缓存,然后再把处理的结果插入到数据库当中去。建立此数据库是为了之后对结果进行归纳,分类整理等处理,图5-2展示了本系统中数据库的图形界面。
5.2 数据可视化
经过以上几个步骤我们大致就将一个粗糙的文件里面的有用的信息全部挖掘,并整理出来了。但是以上挖掘出的数据仅仅只是java里面的object对象,或者是数据库里面存着的一些字段,这些数据对于用户显然是未知的,用户希望看到的是图片或者表格这种直观的表现形式的数据,所以最后还需要把挖掘出的数据通过直观的形式展现给用户。
整体系统采用的是C/S的架构,服务端用来专门处理来自客户端的请求,并返回数据给用户,客户端是发起请求和处理从服务端返回的数据,除此之外客户端也是直接和用户交互的设备,所以数据可视化的工作处理理论上应该由客户端来处理,大概思路就是客户端收到服务端返回的数据之后,通过图形化控件将数据整理,封装。我理解的客户端是只是用来呈现数据的一个软件或者一种手段,客户端软件和平台有关,客户端软件一般和平台有关,比如说Android客户端,IOS客户端,window客户端,Mac客户端等等,各种操作系统的平台都有。Html也是一种解决方案,各个平台的设备上都包含有游览器这个软件,所以只需打开游览器查看服务器返回的数据即可,游览器内核会自己去解析数据里面的各种标签。Html相比其他各个平台的客户端来说,首先html是跨平台的,其次html是不需要下载客户端软件的,直接打开游览器出入地址就行了,简单方便明了。所以本系统也是采取了html的解决方案,通过服务端返回html格式的文本文件给浏览器处理。


本系统采用的是JSP技术,所以每个客户端页面可以看做是一个jsp页面,jsp中包含了html标签页和java代码。一共有两个页面,第一个页面是搜索页面searchpage.jsp,如图5-3所示,这个JSP里面包含了一个搜索框和一个列表,搜索框可以输入关键字和pmid,可以从文档集中搜索出相关的文献,列表里面装有所有搜索出来的文档,如果没有搜索列表里面默认就是文档集中所有的文档,搜索页支持分页的展示如图5-4所示。,然后点击列表里面的一个item文档的话,就会跳转到第二个JSP页面,这个JSP是对应的详情页面,就是展示挖掘出这篇文档的结果,如图5-5和图5-6所示。图中里面展示了整篇文档的pmid,标题,原文,文中的实体,以及实体之间的关系,识别出来的实体会在原文中通过高亮的形式展现出来。实体高亮是通过给文中的实体字符串的两边加上一个特殊的标签,比如在文中被修饰蛋白质实体的两边添加一个类型为蛋白质的标签,然后通过CSS语法指定标签下的文本的背景色,各个实体的具体颜色由标签里面的class属性决定。实体和关系的表格则是通过html5下面标签来实现的。


第六章 总结及展望
6.1 总结
本篇论文主要叙述了文本挖掘技术在生物医学领域上的应用,并且切合实际需求开发了一套有关于蛋白质翻译后磷酸化修饰的信息挖掘系统。随着当今互联网时代信息数据高速增长,文本挖掘技术在处理大量非结构化的数据变得越来越重要,文本挖掘技术在相关领域中的应用也变得越来越重要。
结合当今的文本挖掘技术和实际需求,文本详细叙述了蛋白质翻译后磷酸化修饰的信息挖掘系统的设计与开发。从整个系统工程来看,从需求到设计再到开发,这一流程是严格遵循软件工程中的瀑布流模型的规范。开始首先是通过需求分析获取需求或者是程序的功能,给出具体的需求场景和用例图,然后根据需求来设计整个系统的大体框架,把整体系统分为不同模块,然后依次进行各个模块的开发工作。文本挖掘技术的思想贯穿整个程序,一般的文本挖掘技术的步骤有4个大致步骤:首先是文档预处理,接着是命名实体识别识别,再是知识模型提取,最后是知识模型的评估等等,这对应程序里面的首先是对pubmed文献文件进行预处理,把pubmed文献整理成一个文当集,接着是识别文档里面的文档里面的蛋白质,磷酸化实体等,再是对实体之间关系的提取(知识模型提取),最后是评估。
通过整套系统,生物医学研究者们可以快速获取pubmd文献里的关键信息,比如说被修饰的蛋白质,参与修饰的激酶,修饰位点等等信息,研究者们可以不用通读全文,也能得到这些信息,大大提高了他们阅读文献的效率。对于pubmed文献数据库里面上千万篇的文章,研究者们不可能去用肉眼一一去判别,必须通过计算机去处理得到研究者们想要的东西。
6.2 展望
如今文本挖掘技术已经日趋成熟,并且已经在多个领域已经开始使用,所以文本挖掘技术的准确率和召回率也是却来却多人关注,文本挖掘技术综合了人工智能,机器学习,概率统计学,自然语言处理等各方面的学科,所以我们最希望的就是每一个学科都可以在自己的领域里达到100%的准确率,这些精确度很大一部分取决于机器学习的好和坏,理想情况下,给训练机器训练的东西足够多,训练库也是足够好的,它是能够根据学过的知识给出一个非常满意的判断,当然各个领域都在充实自己机器的训练集,让机器不断学习,来提升准确率,这在未来应该也是一种趋势,因为世界在不断变化,知识、概念、词汇在不断扩展,机器不仅要学会现已有的,更要学会未来人类新增的知识,才能不断成长。此外,未来文本挖掘的重点应该是去从茫茫文本中挖掘出人类未知的知识模型,就好像Marti Hearst说的文本挖掘和信息挖掘最大的区别就是,信息挖掘是你输入一个关键字,挖掘出的信息是你已经预料到的,是已经被发现了的东西,而文本挖掘是输入一个关键字,挖掘出的是你并未知道的知识模型[21]。
结束语
本篇论文主要叙述了文本挖掘技术在生物医学领域上的应用,以及蛋白质磷酸化修饰后信息挖掘系统的具体实现,本系统能够有效的识别出有关于磷酸化信息的文献,并且也能够识别出文本里面的蛋白质实体,激酶实体,和位点实体,效果图可参见图5-5和图5-6,准确率召回率和rlims-p工具本身保持一直,和rlims-p训练语料库的有关。本系统避免了研究人员们人工去查阅文献,大大提高了他们的效率


















 444
444

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











