







Genome assembly and resequencing analyses provide new insights into the evolution, domestication and ornamental traits of crape myrtle

Abstract
Crape myrtle (Lagerstroemia indica) is a globally used ornamental woody plant and is the representative species of Lagerstroemia. However, studies on the evolution and genomic breeding of L. indica have been hindered by the lack of a reference genome. Here we assembled the first high-quality genome of L. indica using PacBio combined with Hi-C scaffolding to anchor the 329.14-Mb genome assembly into 24 pseudochromosomes. We detected a previously undescribed independent whole-genome triplication event occurring 35.5 million years ago in L. indica following its divergence from Punica granatum. After resequencing 73 accessions of Lagerstroemia, the main parents of modern crape myrtle cultivars were found to be L. indica and L. fauriei. During the process of domestication, genetic diversity tended to decrease in many plants, but this was not observed in L. indica. We constructed a high-density genetic linkage map with an average map distance of 0.33 cM. Furthermore, we integrated the results of quantitative trait locus (QTL) using genetic mapping and bulk segregant analysis (BSA), revealing that the major-effect interval controlling internode length (IL) is located on chr1, which contains CDL15, CRG98, and GID1b1 associated with the phytohormone pathways. Analysis of gene expression of the red, purple, and white flower-colour flavonoid pathways revealed that differential expression of multiple genes determined the flower colour of L. indica, with white flowers having the lowest gene expression. In addition, BSA of purple- and green-leaved individuals of populations of L. indica was performed, and the leaf colour loci were mapped to chr12 and chr17. Within these intervals, we identified MYB35, NCED, and KAS1. Our genome assembly provided a foundation for investigating the evolution, population structure, and differentiation of Myrtaceae species and accelerating the molecular breeding of L. indica.
摘要
紫薇(Lagerstroemia indica)是一种全球广泛应用的观赏木本植物,也是千屈菜属(Lagerstroemia)的代表物种。然而,由于缺乏参考基因组,对L. indica的进化和基因组育种研究受到了限制。在本研究中,我们利用PacBio测序结合Hi-C构建,组装了首个高质量的L. indica基因组,并将其329.14 Mb的基因组组装锚定至24条拟染色体。我们发现了一个此前未描述的独立的全基因组三倍化(WGT)事件,该事件发生在L. indica与Punica granatum(石榴)分化之后的3,550万年前。
通过对73份千屈菜属材料进行重测序,我们发现现代紫薇栽培品种的主要亲本是L. indica和L. fauriei。在驯化过程中,许多植物的遗传多样性往往会减少,但在L. indica中并未观察到这一现象。此外,我们构建了一张高密度遗传连锁图,平均图谱距离为0.33 cM。
进一步结合遗传作图和BSA(Bulk Segregant Analysis,混池分析)进行数量性状位点(QTL)分析,发现控制节间长度(IL)的主效区间位于染色体1(chr1),该区域包含CDL15、CRG98和GID1b1基因,它们与植物激素信号通路相关。
在红色、紫色和白色花色的类黄酮代谢途径基因表达分析中,我们发现多个基因的差异表达决定了L. indica的花色,其中白色花的基因表达水平最低。此外,我们对L. indica种群中紫叶和绿叶个体进行了BSA分析,并将叶色相关位点定位至染色体12(chr12)和染色体17(chr17)。在这些区间内,我们鉴定了MYB35、NCED和KAS1等基因。
本研究构建的L. indica基因组为探索桃金娘科(Myrtaceae)物种的进化、种群结构和分化提供了基础,并有助于加速L. indica的分子育种进程。
Introduction引言
Crape myrtle (Lagerstroemia indica), the representative species of the Lagerstroemia genus, is a deciduous shrub or small tree with a long flowering period in summer and is one of the most beloved, iconic trees in tropical and warm-temperate regions. According to the Flora of China, 55 species belong to Lagerstroemia, of which 15 species (eight endemic) are distributed in China [1]. Crape myrtle originated in Southeast Asia to Oceania and began to spread to the Americas and Europe in the late 1700s. China is an important distribution and cultivation centre of L. indica, and has been cultivated there for >1600 years, reaching a prosperous period in the Tang Dynasty [2]. The reason for the popularity of crape myrtle is that it blooms at a time when most trees are not blooming, and it is covered with blooms that will last for months during the hottest part of the summer [1]. In addition to its advantages of unique beauty and aesthetic value, it can resist pollution, absorb harmful gases and dust, and serve as an important landscape plant.
紫薇(Lagerstroemia indica)是千屈菜属(Lagerstroemia)的代表物种,为一种落叶灌木或小乔木,夏季花期较长,是热带和暖温带地区最受喜爱和具有标志性的树种之一。根据《中国植物志》,千屈菜属共有55个物种,其中中国分布有15种(包括8种特有种)[1]。紫薇起源于东南亚至大洋洲,并在18世纪末开始向美洲和欧洲传播。中国是L. indica的重要分布和栽培中心,已有1600多年的栽培历史,并在唐代达到繁盛期[2]。
紫薇之所以备受喜爱,是因为它在大多数树木不开花的时节盛开,并且在最炎热的夏季能够长时间持续开花数月,花朵繁茂、覆盖全树[1]。除了其独特的观赏价值和美学优势外,紫薇还能抗污染、吸收有害气体和粉尘,因此也是重要的园林景观植物。
As early as the middle of the 18th century, crape myrtle was introduced into the southeast of the USA through England. By the early 20th century, it had been widely planted on the east and west coasts of the USA [3]. In the 1960s, Lagerstroemia fauriei, native to Japan, was introduced to America and crossed with Lagerstroemia indica. Hybrids of the two species generally produced excellent offspring. Zhang investigated and collected genetic resources in the Lagerstroemia genus and cultivars in China for the first time [4]. To date, more than 200 hybrid cultivars with diversified plant architectures, different colours, colourful leaves, and strong disease resistance have been successfully bred [2, 5, 6]. In terms of plant architecture, phenotypic and anatomical observations of internodes revealed significant positive correlations between plant height, internode length, and cell number, and internode length was positively regulated by gibberellin [7, 8]. Differentially expressed genes (DEGs) and quantitative trait loci (QTLs) related to the regulation of dwarfism traits in crape myrtle were identified by transcriptomics and QTL mapping [7, 9]. Although the flower colour of crape myrtle is diversified, it still lacks blue, yellow, orange, and green flowers. Flavonoids are considered to be key factors in the determination of petal colour in crape myrtle [10]. In terms of leaf colour, anthocyanins and chlorophylls are considered to be the main determinants of purple and yellow leaf colour, respectively [11, 12]. However, the molecular mechanisms underlying the formation of these traits in L. indica are not clear.
早在18世纪中叶,紫薇通过英国被引入美国东南部。到20世纪初,它已广泛种植于美国东西海岸地区[3]。20世纪60年代,原产于日本的Lagerstroemia fauriei被引入美国,并与Lagerstroemia indica进行杂交,这两个物种的杂交后代通常表现出优良的性状。张氏首次对中国的千屈菜属植物及其栽培品种进行了遗传资源的调查和收集[4]。截至目前,已成功培育出200多个杂交品种,这些品种在植株形态、花色、叶色以及抗病性等方面表现出丰富的多样性[2, 5, 6]。
在植株形态方面,节间的表型和解剖学观察表明,植株高度、节间长度与细胞数目之间存在显著的正相关关系,且节间长度受赤霉素的正向调控[7, 8]。利用转录组学和数量性状位点(QTL)作图,研究者鉴定了与紫薇矮化性状调控相关的差异表达基因(DEGs)和QTLs[7, 9]。
尽管紫薇的花色较为丰富,但仍缺乏蓝色、黄色、橙色和绿色花朵。研究表明,类黄酮是决定紫薇花瓣颜色的关键因素[10]。在叶色方面,花青素和叶绿素分别被认为是紫色叶片和黄色叶片的主要决定因素[11, 12]。然而,L. indica这些性状形成的分子机制仍不清楚。
Over the last 20 years, genomics research in higher plants, especially in Gramineae, Brassicaceae, Orchidaceae, and Rosaceae, has made great advances [13]. With the reduction in sequencing cost, population resequencing based on high-quality genomes can yield a large amount of variation information and multiple types of molecular markers, which are very helpful in the study of population evolution and domestication and for discovering candidate genes associated with target traits based on the genome-wide association study (GWAS) technique [14, 15]. However, the absence of reference genomes for Myrtaceae species has limited our understanding of systematic genomics research. Based on high-quality genomes, new insights can be gained from the analysis of the formation and evolution of important traits. In Myrtales, except for the reports that the genomes of eucalyptus (Eucalyptus grandis) [16], pomegranate (Punica granatum) [17], water caltrop (Trapa natans) [18], clove (Syzygium aromaticum) [19], and other economic tree species have been completed, only the whole set of mangrove genomes is left to explore the evolution process of tropical coastal ecosystems [20]. Whole-genome duplication (WGD), which took place during the evolutionary history of the majority of plant species and offered the potential for new functions and species diversity, could also improve species fitness and resistance. Myrtaceae plants such as Rhodomyrtus tomentosa, E. grandis and P. granatum shared a WGD event from 66.58 to 95.5 million years ago (MYA).
在过去的20年里,高等植物的基因组学研究取得了重大进展,特别是在禾本科(Gramineae)、十字花科(Brassicaceae)、兰科(Orchidaceae)和蔷薇科(Rosaceae)等植物中[13]。随着测序成本的降低,基于高质量基因组的群体重测序能够提供大量的变异信息和多种类型的分子标记,这对于研究种群进化和驯化,以及利用全基因组关联分析(GWAS)技术挖掘与目标性状相关的候选基因具有重要意义[14, 15]。然而,由于桃金娘科(Myrtaceae)植物缺乏参考基因组,限制了我们对其系统基因组学的深入研究。基于高质量基因组的数据分析,可以为重要性状的形成和进化提供新的见解。
在桃金娘目(Myrtales)中,已有研究报道完成了桉树(Eucalyptus grandis)[16]、石榴(Punica granatum)[17]、菱角(Trapa natans)[18]、丁香(Syzygium aromaticum)[19]等经济树种的基因组测序。此外,还完成了一整套红树林基因组研究,以探索热带沿海生态系统的进化过程[20]。
全基因组复制(WGD)在大多数植物物种的进化历史中均有发生,不仅为新功能的产生和物种多样性的形成提供了潜力,还能提高物种的适应性和抗性。研究表明,桃金娘科植物如桃金娘(Rhodomyrtus tomentosa)、E. grandis和P. granatum在6,658万年至9,550万年前(MYA)经历了一次WGD事件。
As L. indica is one of the most representative plants in the Lagerstroemia genus, it is urgent to obtain its genome and systematically conduct functional genomics research. Karyotype analysis with 45S rDNA-FISH showed that the chromosomes of Lagerstroemia species are small and numerous (2n = 2x = 48), consistent with the results of flow cytometry of 10 species of Lagerstroemia (341.00 ± 2.00–370.00 ± 8.89 Mb) [21, 22]. Due to the lack of a reference genome, very large datasets cannot be effectively integrated and utilized, which seriously hinders research on the evolution, domestication, and molecular breeding design of crape myrtle and Lagerstroemia species.
由于L. indica是千屈菜属(Lagerstroemia)中最具代表性的植物之一,迫切需要获得其基因组并系统开展功能基因组学研究。利用45S rDNA荧光原位杂交(FISH)进行核型分析表明,千屈菜属植物的染色体数量多且较小(2n = 2x = 48),这一结果与对10种千屈菜属植物的流式细胞术分析结果一致(基因组大小范围为341.00 ± 2.00 Mb 至 370.00 ± 8.89 Mb)[21, 22]。由于缺乏参考基因组,大规模数据集无法得到有效整合和利用,这严重阻碍了对紫薇及千屈菜属植物的进化、驯化以及分子育种设计的研究。
Here we obtained the chromosome-level genome of L. indica by using PacBio and Hi-C technology, performed genome resequencing and evolutionary analysis of 73 closely related species and cultivars, constructed a high-density genetic linkage map by resequencing and QTL mapping for plant height and revealed comprehensive models of plant architecture, petal colour, and leaf colour by multi-omics. This study will provide an important platform for genetic breeding and ornamental trait improvement in L. indica.
在本研究中,我们利用PacBio和Hi-C技术获得了荷花(L. indica)的染色体水平基因组,对73种密切相关物种和品种进行了基因组重测序和进化分析,通过重测序和QTL定位构建了高密度遗传连锁图谱,并针对植物株高进行了研究,同时通过多组学手段揭示了植物株型、花瓣颜色和叶片颜色的综合模型。这项研究将为荷花的遗传育种和观赏性状改良提供重要的平台。
Results
Chromosome-scale reference genome assembly of L. indica
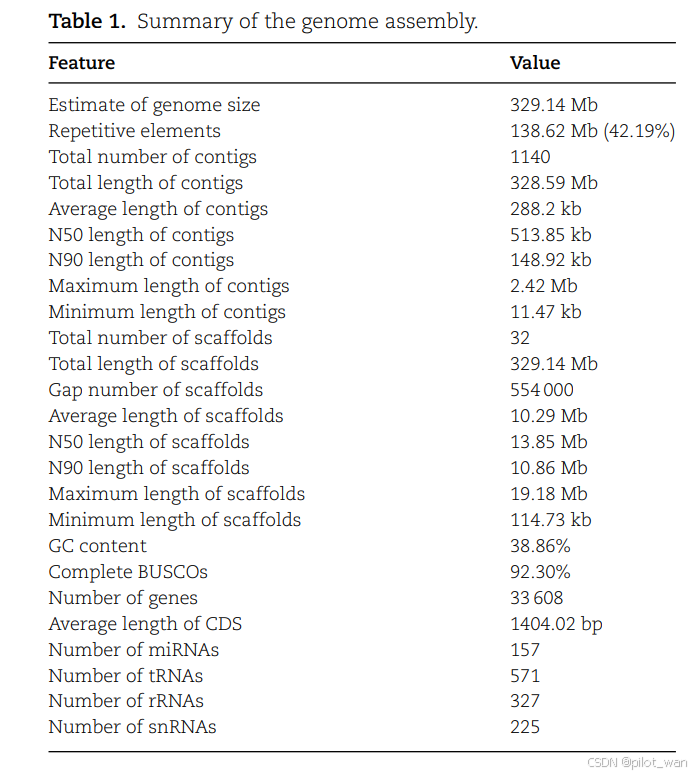
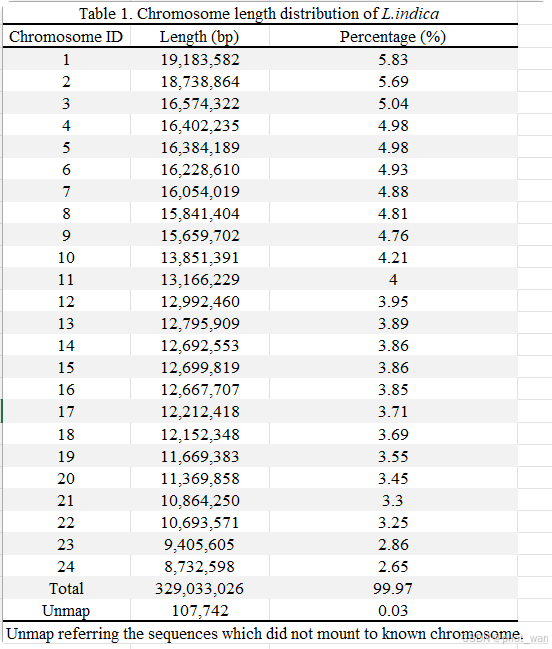
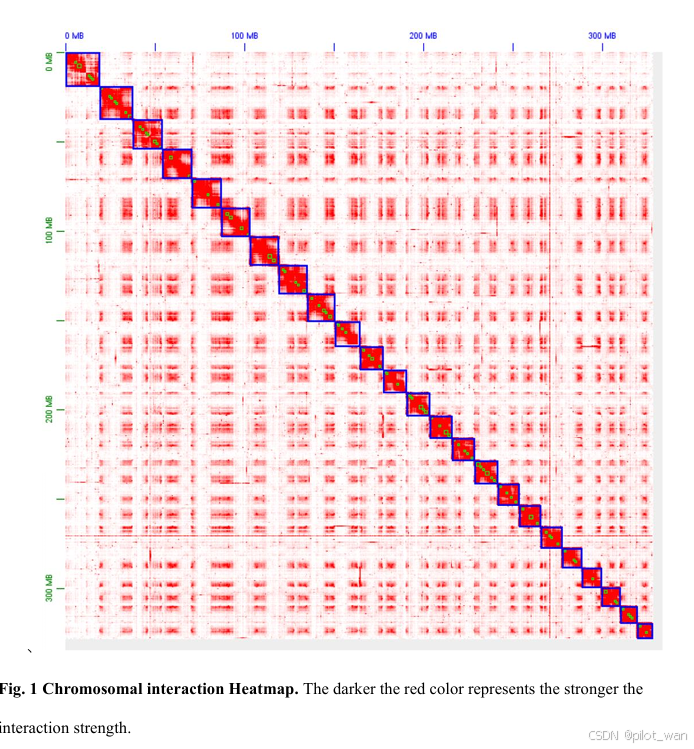
A diploid (2n = 2x = 48) of L. indica was used for whole-genome sequencing and chromosome-level assembly with PacBio sequencing and Hi-C technologies, respectively. We obtained 100× coverage of PacBio long-read sequencing data (33.15 Gb) and 112× coverage of Hi-C paired-end reads (37.1 Gb). The complete genome assembly size of L. indica was 329.14 Mb with a scaffold N50 of 13.85 Mb. The genome was assembled into 24 chromosomes, and the percentage of sequences anchored to chromosomes was 99.97%. Detailed genome assembly statistics and the chromosome-scale scaffold length range are shown in Fig. 1, Table 1, and Supplementary Data Table S1.
我们利用PacBio测序技术和Hi-C技术,分别对荷花(L. indica)的二倍体(2n=2x=48)进行了全基因组测序和染色体水平组装。我们获得了100倍覆盖率的PacBio长读长测序数据(33.15 Gb)和112倍覆盖率的Hi-C配对末端读取数据(37.1 Gb)。荷花的完整基因组组装大小为329.14 Mb,scaffold N50为13.85 Mb。基因组被组装成24条染色体,锚定到染色体上的序列占比为99.97%。详细的基因组组装统计数据以及染色体水平scaffold长度范围如图1、表1和补充数据表S1所示。
图1
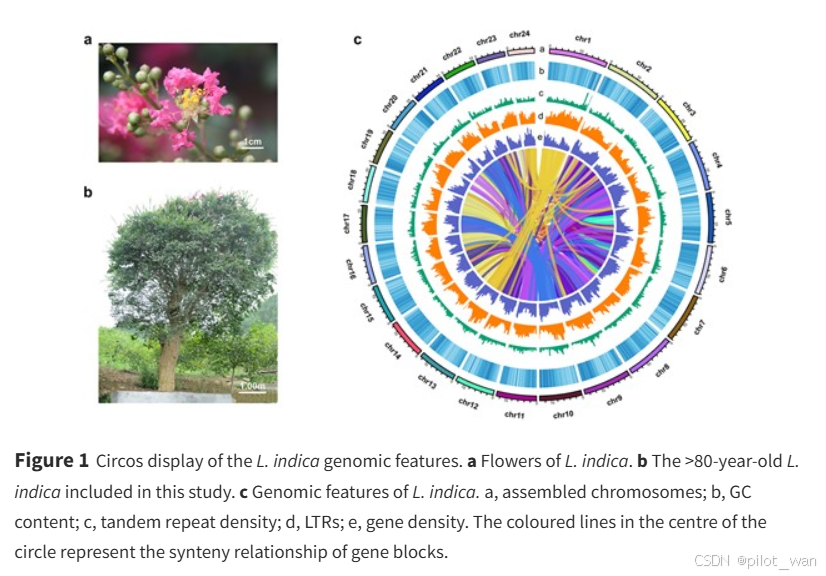
图1 荷花(L. indica)基因组特征的Circos展示。a. 荷花的花朵。b. 本研究中包含的80多年历史的荷花。c. 荷花的基因组特征。a,组装的染色体;b,GC含量;c,串联重复序列密度;d,长末端重复序列(LTRs);e,基因密度。圆圈中心的彩色线条表示基因块的同源关系。
表1
数据表S1
基因组注释Genome annotation
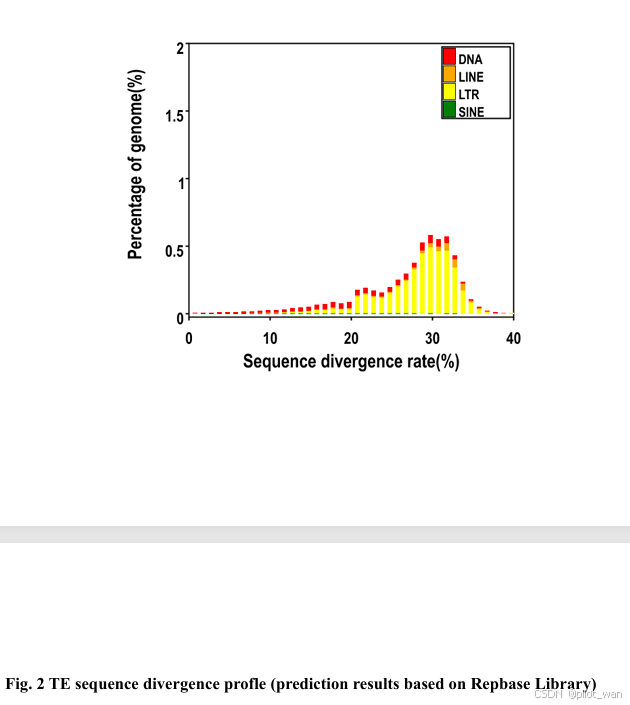
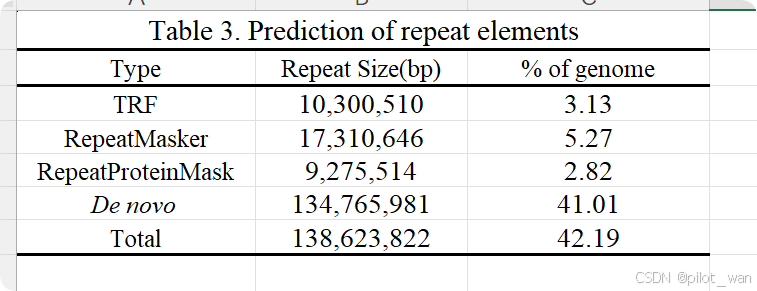
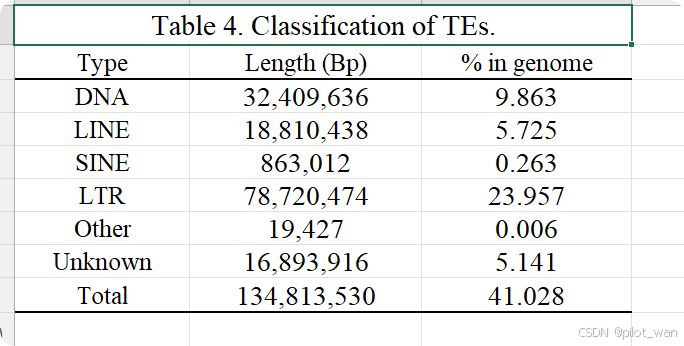
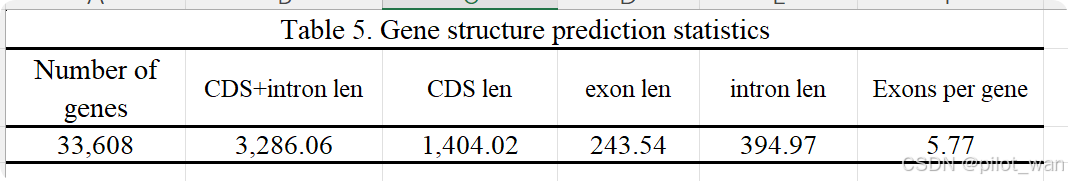
Overall, ~138.62 Mb of genome sequences in L. indica were identified as repetitive elements by the repeat annotation processes mentioned above and accounted for ~42.19% of the whole genome. The detailed prediction resources and classification of TEs are listed in Supplementary Data Fig. S2, Supplementary Data Tables S3 and S4. We identified 33 608 genes in L. indica, with an average coding sequence (CDS) length of 1.4 kb (Supplementary Data Table S5, Supplementary Data Fig. S3), and the BUSCO evaluation of the annotated protein sequences was 93.4%. A total of 31 487 genes were functionally annotated in L. indica, accounting for 93.69% of all predicted genes (Supplementary Data Table S6). The non-coding RNAs (miRNAs, tRNAs, rRNAs and snRNAs) were also annotated and are presented in Supplementary Data Table S7.
总体而言,通过上述重复序列注释流程,荷花(L. indica)的基因组序列中有约138.62 Mb被鉴定为重复元件,占整个基因组的约42.19%。详细的转座元件(TEs)预测资源和分类信息见补充数据图S2、补充数据表S3和S4。我们在荷花中鉴定出33,608个基因,平均编码序列(CDS)长度为1.4 kb(补充数据表S5,补充数据图S3),其注释蛋白序列的BUSCO评估值为93.4%。在荷花中共有31,487个基因被进行了功能注释,占所有预测基因的93.69%(补充数据表S6)。此外,非编码RNA(包括miRNA、tRNA、rRNA和snRNA)也进行了注释,相关信息见补充数据表S7。
图S2 图2 转座元件(TE)序列差异分析图谱(基于Repbase数据库的预测结果)。
补充数据表S3和S4
补充数据表S5
补充数据图S3
补充数据表S6
补充数据表S7
比较基因组学与进化分析Comparative genomic and evolutionary analysis
The 17 species contained a total of 33 875 gene families, of which 16 126 gene families (29 357 genes) were found in L. indica (Supplementary Data Table S8, Supplementary Data Fig. S4). Clustering of gene families from four species (L. indica, P. granatum, Arabidopsis thaliana and Carica papaya) revealed that 9675 genes were common to these species, whereas 3572 genes were unique to L. indica (Supplementary Data Fig. S5).
这17个物种共包含33,875个基因家族,其中16,126个基因家族(包含29,357个基因)存在于荷花(L. indica)中(补充数据表S8,补充数据图S4)。对4个物种(荷花[L. indica]、石榴[P. granatum]、拟南芥[Arabidopsis thaliana]和木瓜[Carica papaya])的基因家族进行聚类分析显示,9675个基因是这些物种共有的,而3572个基因是荷花(L. indica)所特有的(补充数据图S5)。
补充数据表S8
补充数据图S4

补充数据图S5
图5 比较了四种植物(荷花[L. indica]、梅花[P. mume]、拟南芥[A. thaliana]、木瓜[C. papaya])的共有基因家族和特
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









