






原文标题:Enhanced Integration ofSingle-Cell Multi-Omics Data Using Graph Attention Networks
跨不同组学层的数据集成面临的挑战:高维度、异质性和稀疏性。
变分自编码器(Variational Autoencoder,VAE)是一个非对称的深度学习框架,它由三个主要部分组成:编码器、隐藏层和解码器。编码器学习输入数据的低维表示,而隐藏层控制这些特征的分布,使模型能够捕获复杂的数据结构。然后,解码器从这些潜在表示中重建原始输入数据。
高斯混合模型(Gaussian Mixture Model,GMM)是由多个高斯分布组成的模型,其密度函数为多个高斯密度函数的加权组合。
解决VAE的方法在多组学数据分析中的局限性,开发了深度典型相关分析(DCCA)和单细胞多组学差异推断(scMVP)方法。
DCCA使用多个VAE,每个VAE专门用于处理不同类型的组学数据。该架构允许DCCA捕获每个数据集的独特特征,同时利用相互监督来识别和建模不同组学数据类型嵌入之间的相关性。有助于更全面地了解各个生物层之间的关系。
scMVP采用了多模态的VAE方法,结合了针对不同类型的多组学数据的特定的编码器和解码器。这种设计使scMVP能够有效地捕获每个数据集的细微差别,同时也利用了包含注意力机制的联合潜在空间。注意力机制允许模型在不同的组学层中专注于最相关的特征,从而增强了其识别有意义的生物信号和相互作用的能力。
作者提出的scMGAT (基于多头图注意力网络的单细胞多组学数据分析)方法在单细胞多模态组学分析的背景下引入了一种整合转录组学和表观基因组学数据的创新方法,scMGAT使用多头注意力机制来有效协调和优化不同类型组学数据之间交换的可靠信息,从而解决单细胞多模态组学数据集中存在的固有异质性问题。在评估不同组学数据类型之间的相似性时,引入了自注意力机制,提高了模型的处理效率。实验结果表明,scMGAT能够从不同的单细胞组学层中提取有意义的特征,并有效地识别这些不同类型的组学数据之间的重要关联。
方法
scMGAT生成模型
ScMGAT采用了一种集成自编码器( AEs )和图注意力网络( GATs )的新方法来有效地分析单细胞多组学分析数据,分为三个关键步骤。
在第一步中,两个组学数据集经过必要的预处理,包括零值基因过滤和对数转换,以提高数据质量(图1A )。随后,AE模块通过最小化均方误差作为损失函数对组学数据进行优化和合并,从而使数据具有更鲁棒的表示。
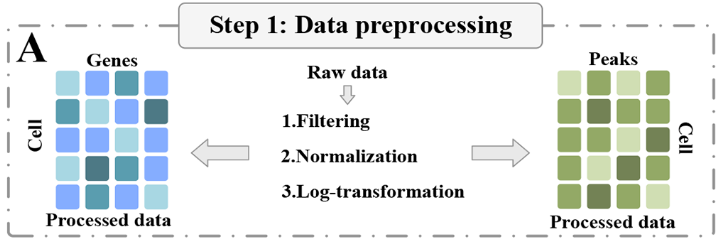
原始数据进行过滤、标准化、对数变换等预处理。
第二步利用从AE模块得到的隐层表示在GAT框架下构造邻接矩阵。这个矩阵便于建立连接单细胞多组学分析数据的各个组成部分的融合嵌入。GAT中的图卷积层结合了多头注意力机制,能够很好地管理节点与其邻居之间的关系,从而增强模型捕获复杂交互的能力(图1B )。
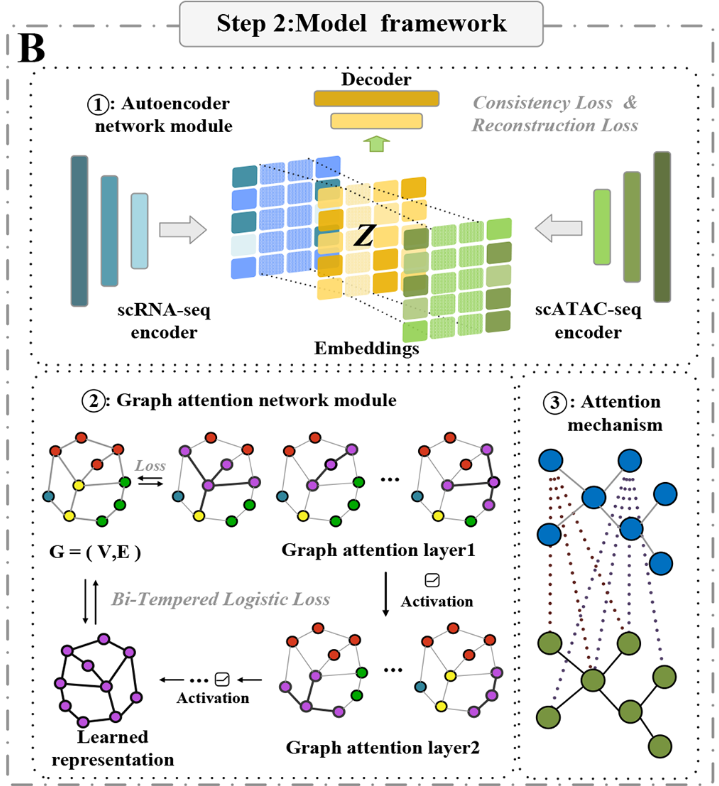
嵌入层数据构建邻接矩阵并输入到图注意力网络中,产生图输出层作为指定维度的输出数据。
在数据预处理过程中,仅保留具有重叠组学矩阵的细胞。原始数据作为多组学数据输入,以保证准确的信息借用。图中的每个节点代表单个细胞,具体来说是细胞在融合组学特征空间中的低维嵌入。
边缘表示细胞之间的相似性或关系,基于这些融合特征计算。
第三步,利用sc MGAT产生的输出进行深入的基因水平分析,为下游的研究和应用奠定基础(图1C )。这个全面的框架使我们能够更深入地理解多组学数据集背后复杂的生物学过程。
自编码网络模块
自编码器( AE )是一种无监督的多层前馈神经网络,旨在通过最小化重构输出和原始输入数据之间的损失来学习数据特征。AE是通过编码器-解码器架构实现的,其中编码器将输入数据映射到低维隐空间,解码器从这种隐表示中重构数据。AE的主要目标是提取相关特征,同时保留关键信息。在scMGAT模型中,AE作为无监督学习的关键组件,促进了多组学数据在嵌入层的融合。
scMGAT模型的融合过程包括两个关键步骤。首先,通过零值基因过滤和对数变换等技术获得两个组学数据集的预处理矩阵,并将其独立地输入到各自的编码器中。这种编码过程将组学数据转化为低维表示,捕获了输入的本质特征,有效地减少了噪声和冗余。其次,将从编码器中提取的隐藏层进行融合——通常是通过级联或元素级操作来创建嵌入层。该嵌入层对整合的多组学信息进行编码,从而为后续的分析任务提供便利。
自编码网络模块由编码器和解码器组成。编码器由多层组成,处理来自每个组学类型的输入数据,捕获基本特征。解码器利用这种潜在表示对原始多组学数据进行重构,保证重构误差最小。对于scRNA - seq数据X1∈R m×n1和scATAC - seq数据X2∈R m×n2,其中m为样本数量,n1,n2分别为特征数量;每个组学类型的数据通过多个隐藏层进行处理。每一层的变换都可以用下面的数学表达式来表示:
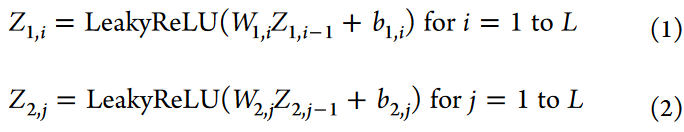
每一层之间使用的激活函数是LeakyReLU,它引入了非线性,使模型能够学习数据内部的复杂模式和关系。该函数的数学表达式为:

合并后的数据Z是将两个数据集的最后一层表示串联起来得到的:

从潜在空间Z重构原始数据如下:
![]()
然后将重建后的数据X^映射回原始的数据维度:

采用均方误差( Mean Squared Error,MSE )损失函数计算重构误差:
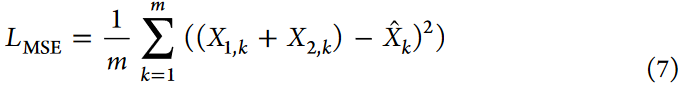
嵌入层的多组学数据融合是通过对编码器得到的隐含层进行融合,并应用融合操作(例如,拼接或元素加法)对表示进行组合,然后将融合后的层作为输入进行下游分析的过程实现的。这种融合方法允许scMGAT模型利用来自多个组学数据源的互补信息,捕获潜在的相互依赖关系。在训练过程中,自编码器( autoencoder,AE )模型通过使用MSE损失函数最小化输出与原始输入之间的重构损失。MSE损失量化了重建数据点与原始数据点之间的平均平方差。通过迭代调整模型参数——包括编码权重矩阵( Wi )和偏置向量( bi ),AE优化网络实现精确的数据重建。
图注意力网络模块
在训练自编码器( AE )的基础上,scMGAT模型根据学习到的多组学数据表示构建邻接矩阵,记为G。随后,使用图注意力网络( Graph Attention Network,GAT )算法和自注意力层来识别G中更有代表性的节点,并评估各个节点在各自邻域内的相关性。这种方法使得自感知层节点能够关注G中邻居节点的特征,从而有利于信息的全面融合。该模型使用的注意力机制为每个节点分配不同的权重,使其在处理局部数据的同时强调全局信息。该机制通过考虑不同节点之间的重要性和相互关系,增强了单细胞多组学数据的整合。通过利用自注意力机制,该模型有效地捕获和组合了来自各种组学来源的相关信息,从而增强了单细胞多组学数据的表示和融合。
GAT模块接收一组细胞特征作为输入,表示为h = { h1,h2,· · ·,hN },其中hi∈RF表示N个细胞,每个细胞用F个特征值表征。该模块产生新的、鲁棒性更强的特征作为输出,记为h′= { h1′,h2′,· · ·,hN′},其中hi′∈RF′,F′表示新特征向量的长度。原始特征到增强特征的转换在每个细胞节点处通过权重矩阵R(F′× F)进行参数化。为了融入自注意力机制,引入共享函数a:RF′× RF′→R。该函数结合了单元节点i和j的加权输出(Whi,Whj),得到一个注意力系数eij = a(Whi,Whj)。自注意力机制允许模型中的每个节点与其他节点进行交互,从而捕获节点j的特征值对节点i的重要性。为了确保计算只考虑i的邻居节点,其中j∈Ni,采用了隐藏注意力技术。这种技术限制了对图中邻接点的关注,从而排除了不相邻的节点。利用softmax方法对原始注意力得分eij进行处理,得到注意力系数αij,保证所有相邻节点的注意力得分归一化,且其权重和为1。这可以表示为:
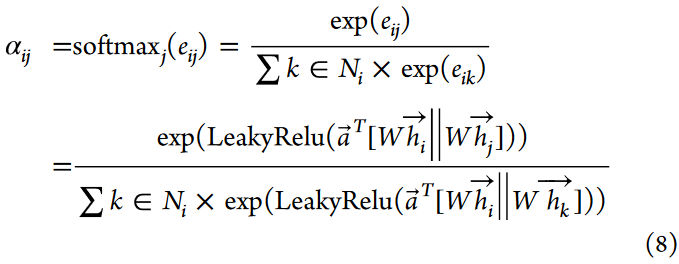
该规定便于在生成输出特征的线性组合计算中确定每个节点的特征重要度。然后将LeakyReLU激活函数应用到线性组合中,表示为LeakyRelu ( aT [ Whi∥Whj ] ),其中T表示转置,aT表示可学习的权重向量,∥表示级联操作。这一过程增强了细胞间的生物变异,稳定了自学习机制的效果。具体来说,该函数被执行多次以产生线性输出,然后将得到的特征进行连接和平均。具体公式如下:
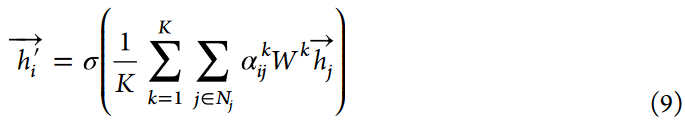
节点ii的更新特征记为hi′,通过以下步骤计算:
(1)应用sigmoid激活函数σ生成更新的特征向量hi′;
(2)每个多传感头注意力头k的输出,用α ij k表示,与相邻节点( j∈Nj)的特征向量hj进行逐元素相乘;
(3)将这些相乘后的值在所有相邻节点( j∈Nj)上求和;
(4)所得和乘以第k个头部的线性变换矩阵Wk;
(5)使用1/K作为尺度因子,计算所有K多传感头注意力的转换和的平均值。
该模型通过引入多头注意力机制,有效地整合了多组学数据中的有价值信息。这种方法便于对隐藏层内的数据进行无害化处理,从而得到非常有利于下游分析的输出结果。图注意力网络( Graph Attention Network,GAT )模型包含连接层,每个连接层都配备了注意力机制。在初始层中,节点获取输入数据的特征维度。后续的连接层旨在增强模型捕捉数据内部复杂关系的能力。图注意力连接层之间使用的激活函数是整流线性单元( ReLU ),它引入了非线性,使模型能够捕获数据中错综复杂的模式。
为了优化性能,使用Adam优化器,权重衰减为1 × 10-3。这种优化技术的结合提高了训练效率,缓解了过拟合,从而提高了泛化能力和更稳健的结果。为了进一步提高scMGAT模型的准确性,并降低数据中隐藏噪声的负面影响,采用基于Bregman散度的双向logistic损失算法作为损失函数。该方法解决了实验过程中未知噪声对重要信息选取的影响,使得模型精度得到显著提高。在scMGAT模型中使用的损失函数定义如下:
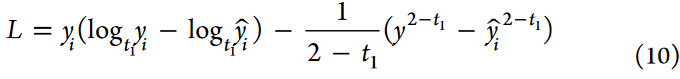
式中,yi和^yi分别表示来自图注意力层的目标和预测值。通过对输入单元数据的对数变换和指数函数对输入的调整来缓解异常噪声对模型性能的影响。值得注意的是,当t1和t2取值为1时,scMGAT模型中使用的损失函数等价于激活函数中的KL散度。
结果
细胞聚类性能的评价指标
在这项研究中,使用调整兰德指数( ARI )和标准化互信息( NMI )来评估scMGAT模型和其他可比方法在真实数据集上的聚类性能,包括可用的和自标注的标签。
调整兰德指数( ARI )
ARI度量了真实标签和从模型中得到的预测聚类结果之间的相似性。ARI的计算公式如下:

兰德指数( Rand Index )量化了两组数据之间的相似性,同时考虑了聚类结果和真实标签。ARI的取值范围为[ 0、1 ],其值越大,表明聚类性能越好。ARI值为0表示完全随机的聚类结果,ARI值为1表示与真实聚类标签完全匹配。
标准化互信息( NMI )
NMI量化了聚类结果与真实标签之间的信息共享程度,衡量了它们之间的互信息。虽然ARI强调聚类中的结构相似性,但NMI提供了更稳健的评估,特别是在不平衡聚类的情况下。NMI的计算公式如下:
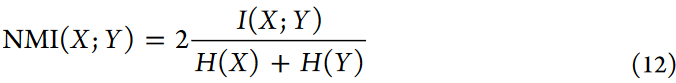
NMI得分越高,表明评价方法的聚类性能越好。该公式融合了互信息I和熵函数H,它们量化了X和Y之间共享的信息水平。具体地,H(X)度量了真实标签中的不确定性,而H(Y)度量了预测标签中的不确定性。
在cellMix和小鼠肾脏数据集上进行了scMGAT的分析
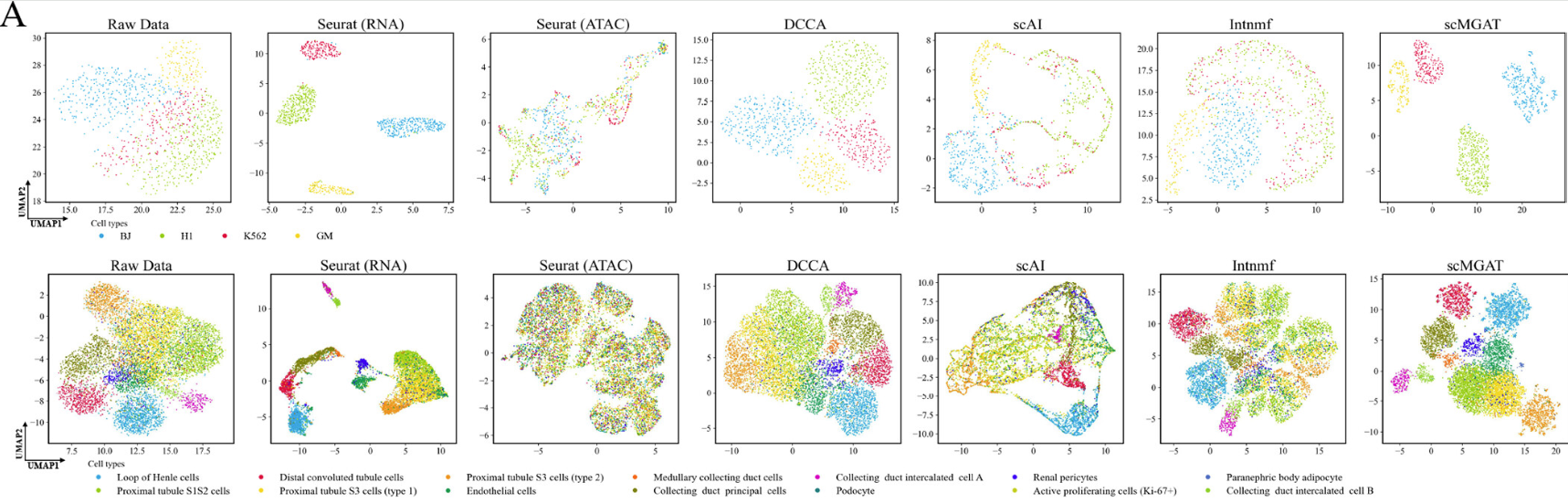
每个子图中的细胞根据其对应的细胞类型进行颜色编码。
使用调整兰德指数( ARI )和标准化互信息( NMI )值评估的聚类精度进一步支持了scMGAT (图A)的优越性能。Intnmf在多组学数据中表现出比scAI更差的性能。
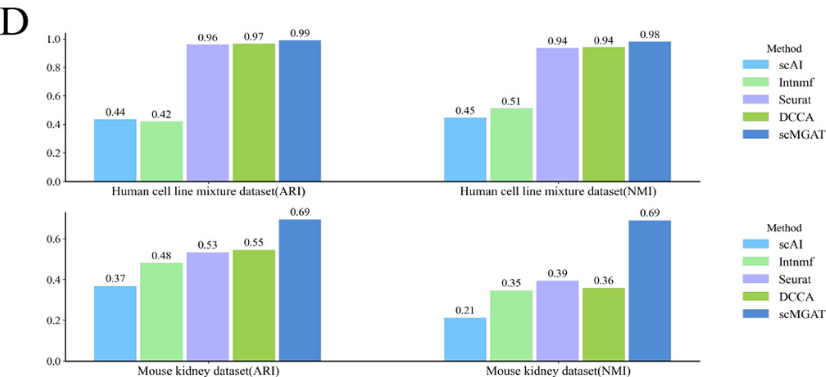
评估各种方法在cellMix和小鼠肾脏数据集上的聚类精度。
分析发现scMGAT的多传感头注意机制有效地同时包含了细胞和基因(图D)。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









