




引言
空间组学技术(如 Xenium、Visium 等)正在彻底改变我们对生物组织的理解,为我们提供了基因表达和空间位置的双重信息。然而,处理这些多维、异构的数据仍然是一个巨大的挑战。为了解决这一问题,SpatialData 应运而生——一个开源的、通用的数据框架,旨在统一存储、处理和分析空间组学数据。

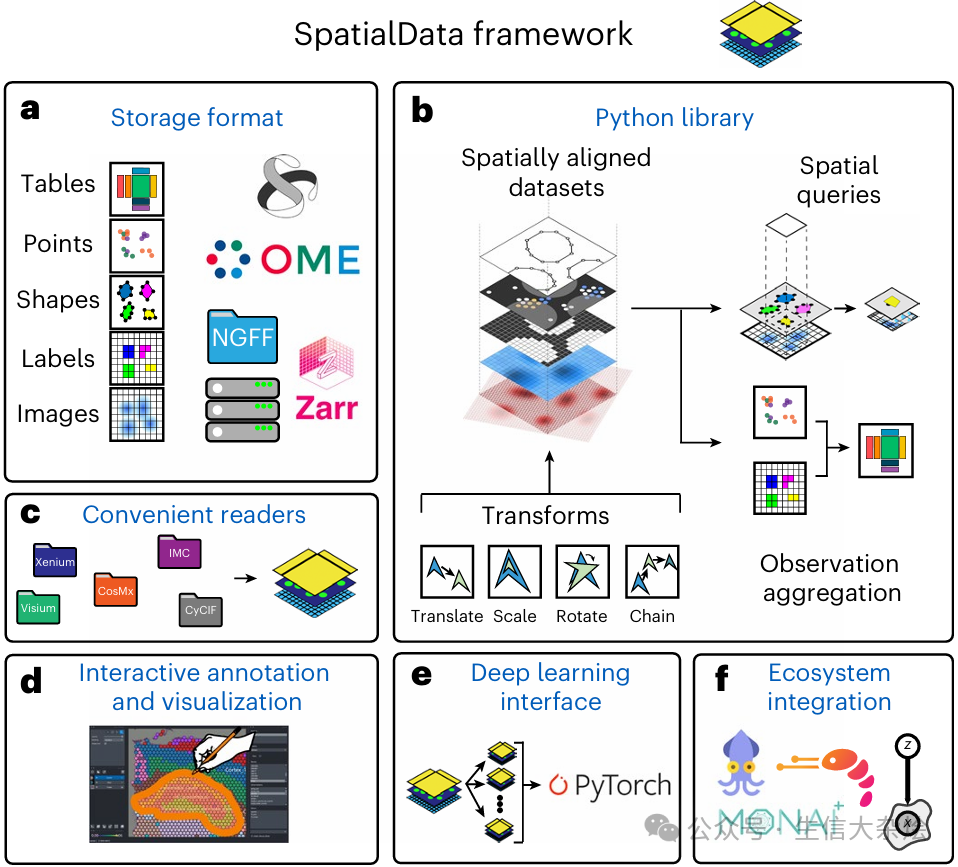
目录
-
什么是 SpatialData?
-
SpatialData 的核心设计
-
SpatialData 的主要功能
-
实战:使用 SpatialData 读取单、多样本 Xenium 数据
1. 什么是 SpatialData?
SpatialData 是一个开源的、多平台兼容的空间组学数据框架,旨在解决以下问题:
- 数据异构性
不同的空间组学技术(如 Xenium、Visium、CyCIF 等)生成的数据类型和格式各异。
- 数据量大
图像和转录本数据通常占用数百 GB 的存储空间。
- 空间对齐
不同数据模态的空间分辨率和区域采集范围通常不一致,需要对齐到统一的坐标系。
- 跨模态分析
整合多模态数据(如基因表达、形态学图像)需要灵活的工具支持。
SpatialData 提供了一种统一的数据格式和 Python 库,支持数据加载、对齐、查询和跨模态分析,极大地简化了空间组学数据的处理流程。
2. SpatialData 的核心设计
SpatialData 的核心设计包括以下几个方面:
(1) 数据格式
SpatialData 基于 OME-NGFF 和 Zarr 文件格式,支持以下五种基本数据类型:
- Images
光栅图像(如 H&E 染色图像)。
- Labels
分割掩模(如细胞和核的分割结果)。
- Points
点数据(如单分子转录本的位置)。
- Shapes
几何形状(如多边形的感兴趣区域)。
- Tables
表格数据(如基因表达矩阵和注释信息)。
这种格式支持云存储和延迟加载,适合处理大规模数据。
(2) Python 库
SpatialData 提供了 Python 库,支持以下功能:
- 延迟加载
支持处理超出内存大小的数据。
- 空间对齐
通过坐标变换将不同模态的数据对齐到统一坐标系。
- 数据查询与聚合
支持按空间区域查询和跨模态数据聚合。
- 生态系统集成
与 Scanpy、Squidpy 等单细胞分析工具无缝集成。
(3) 可视化工具
SpatialData 提供了 napari-spatialdata 插件,支持交互式数据探索和注释,如绘制感兴趣区域和定义对齐标志点。
3. SpatialData 的主要功能
以下是 SpatialData 的一些核心功能:
(1) 数据加载与对齐
SpatialData 支持从多种空间组学技术加载数据,并通过坐标变换将它们对齐到统一坐标系。例如,可以将 Xenium 和 Visium 数据对齐到同一组织切片。
(2) 数据查询与聚合
SpatialData 支持按空间区域查询数据,并提供了灵活的聚合功能。例如,可以将单分子转录本聚合到细胞或 Visium 捕获点中。
(3) 交互式注释
通过 napari-spatialdata 插件,用户可以交互式地定义空间注释(如感兴趣区域),并将其用于后续分析。
(4) 生态系统集成
SpatialData 与 scverse 生态系统(如 Scanpy、Squidpy)无缝集成,支持单细胞和空间组学数据的联合分析。
4. 实战:使用 SpatialData 读取多样本 Xenium 数据
spatialdata github地址:https://github.com/scverse/spatialdata
(1) 安装 SpatialData
首先,安装 SpatialData 及其依赖库(安装最新版本,之前的版本有几个功能函数有bug):
pip install "spatialdata[extra]"
(2) 单个样本数据读取 (10x Breast Cancer FFPE Xenium5k demo data)
import scanpy as scimport spatialdata_plotfrom spatialdata_io import xeniumfrom spatialdata_io.experimental import to_legacy_anndataimport matplotlib.pyplot as pltfrom matplotlib.pyplot import rc_contextsdata = xenium(path='./Xenium_Prime_Breast_Cancer_FFPE_outs',cells_boundaries=True,nucleus_boundaries=True,cells_as_circles=True,aligned_images=True,n_jobs=8,)sdata
spatialData对象包含的内容
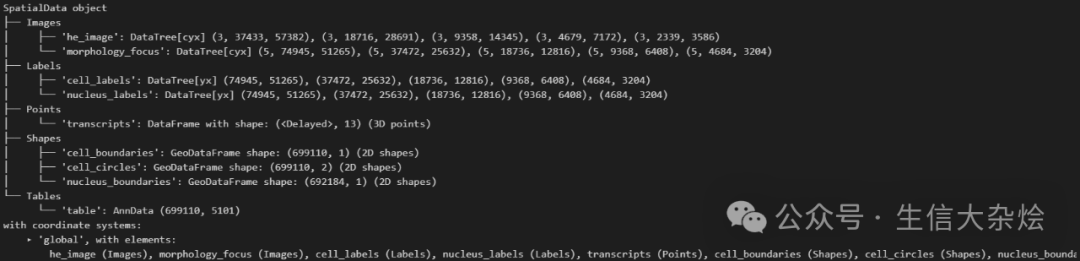
单细胞数据
adata = to_legacy_anndata(sdata, include_images=True, table_name='table')

adata使用scanpy绘图
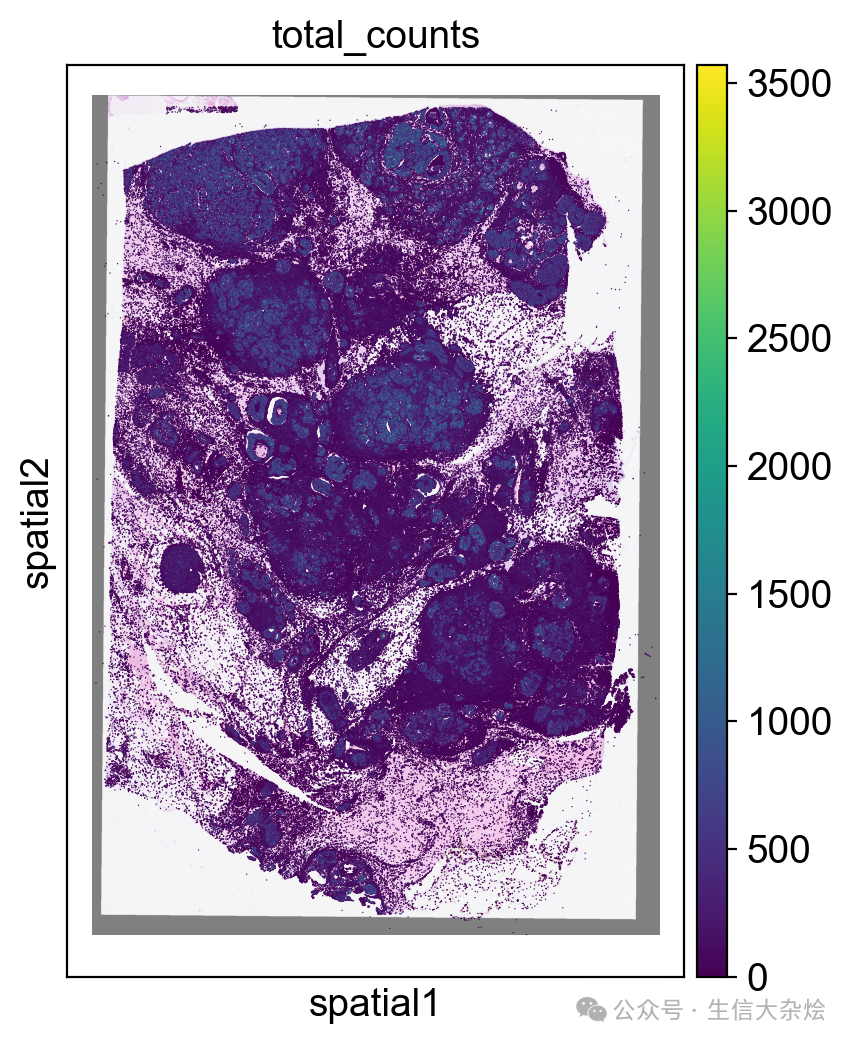
spatialdata-plot简单绘图功能
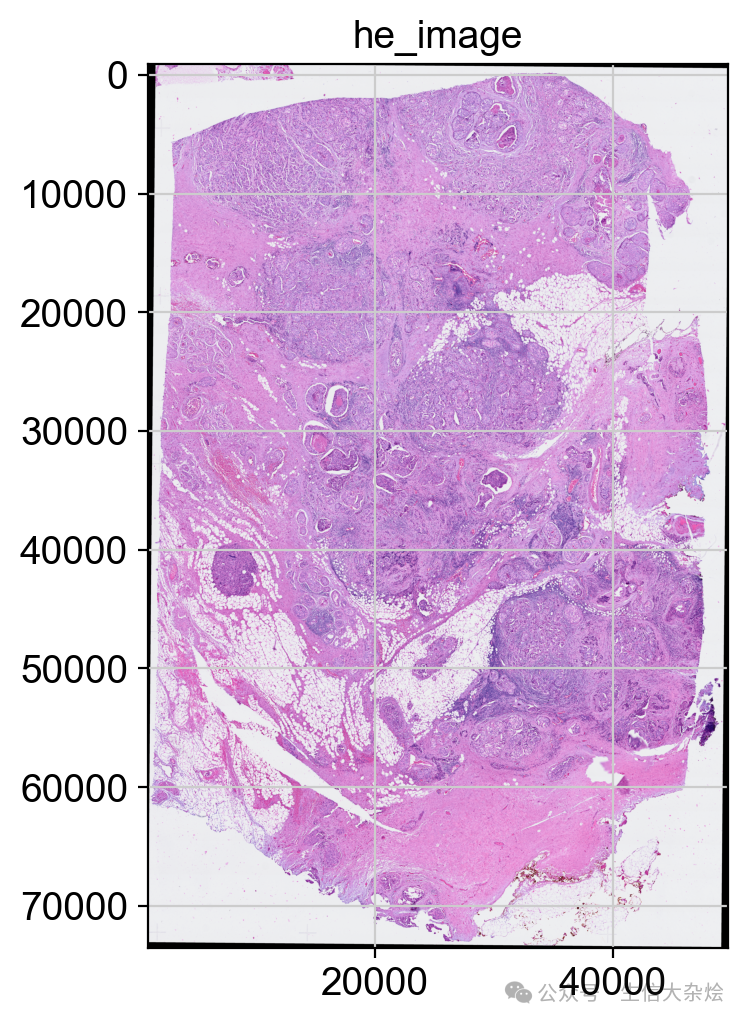
sdata.pl.render_images("he_image").pl.show(title="he_image")
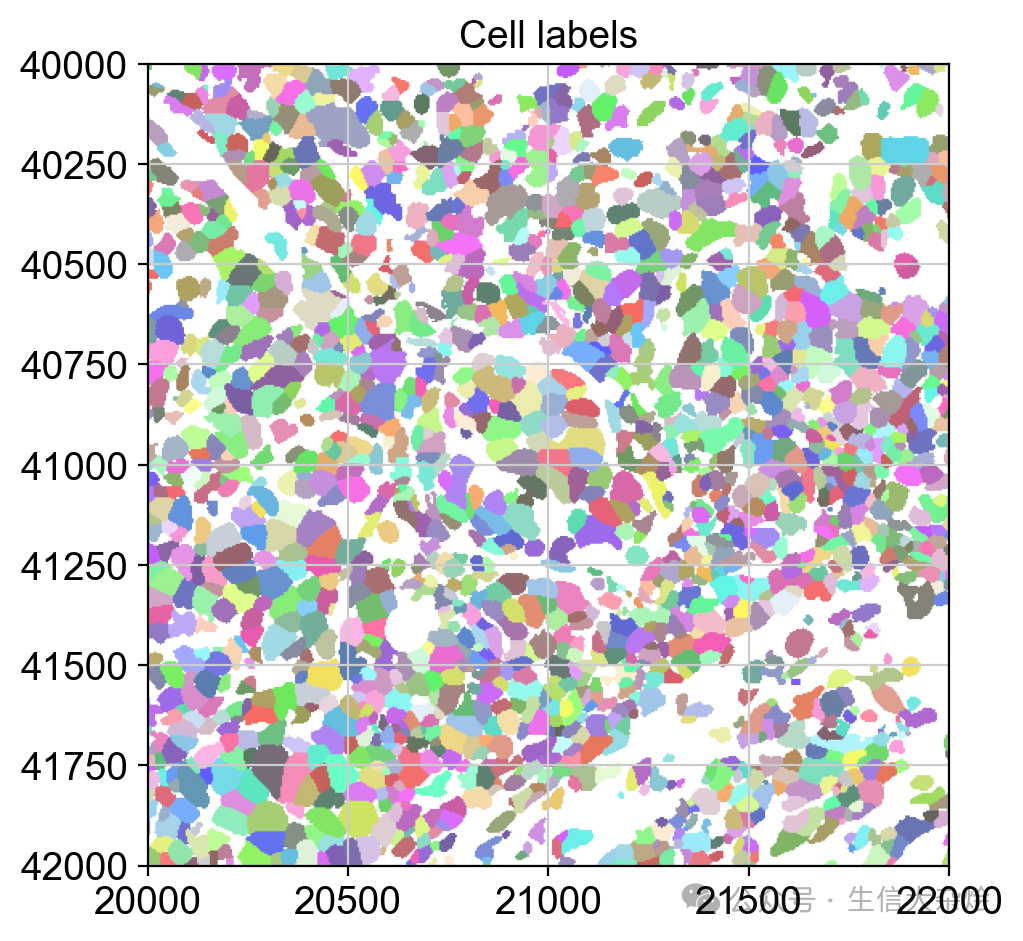
from spatialdata import bounding_box_querycrop0 = lambda x: bounding_box_query(x,min_coordinate=[20000, 40000],max_coordinate=[22000, 42000],axes=("x", "y"),target_coordinate_system="global",)crop0(sdata).pl.render_labels("cell_labels").pl.show(title="Cell labels", coordinate_systems="global")
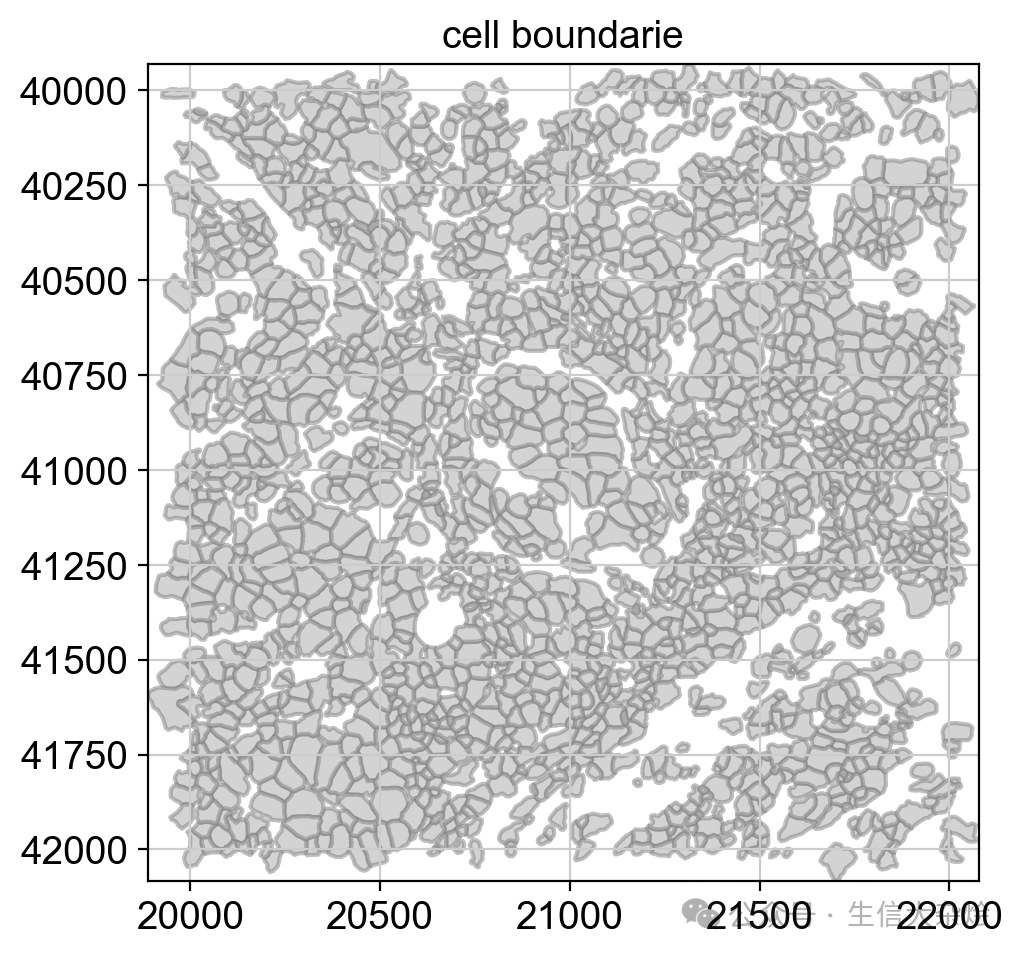
crop0(sdata).pl.render_shapes("cell_boundaries", outline_color='grey', outline_alpha=0.5).pl.show(title="cell boundarie", coordinate_systems="global")
(3) Xenium5k多样本并行读取
import pandas as pdimport scanpy as scimport multiprocessingimport spatialdata_plotimport spatialdata as sdfrom spatialdata_io import xeniumfrom spatialdata_io.experimental import to_legacy_anndataimport matplotlib.pyplot as pltfrom matplotlib.pyplot import rc_contextimport warningswarnings.filterwarnings('ignore')
def load_xenium_data(data_dir, sample_info):def sd_read_xenium(raw_dir, sample_name, sample_group, result_list):sdata = xenium(raw_dir, cells_as_circles=True, n_jobs=6)new_images = {f"{name}_{sample_name}":img for name, img in sdata.images.items()}sdata.images = new_imagesif hasattr(sdata, 'tables'):for name, table in sdata.tables.items():table.obs['sample'] = sample_nametable.obs['group'] = sample_groupresult_list.append(table)with multiprocessing.Manager() as manager:result_list = manager.list()processes = []with open(sample_info, 'r') as f:for line in f.readlines():line = line.strip().split('\t')raw_name = line[0]sample_name = line[1]sample_group = line[2]p = multiprocessing.Process(target=sd_read_xenium,args=(os.path.join(data_dir, raw_name), sample_name, sample_group, result_list,))processes.append(p)p.start()for p in processes:p.join()adata = result_list[0].concatenate(result_list[1:], index_unique=None)adata.obs_names_make_unique()adata.var_names_make_unique()del adata.obs['batch']adata.obs.reset_index(drop=True, inplace=True)return adata

















 492
492

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









