




前面一节经过基础分析后,我们得到了8um bin单细胞降维聚类结果,接下来就可以对上述降维聚类后的结果进行细胞类型注释了,细胞类型注释不管单细胞还是空间转录组都是非常重要的一步,如果对于细胞类型注释不准确,后面的分析基本上无从说起了。
这里我们使用文章中提到的,基于单细胞数据进行解卷积注释,文章中用到的方法是RCTD(Robust decomposition of cell type mixtures in spatial transcriptomics) https://www.nature.com/articles/s41587-021-00830-w 使用单细胞数据作为参考来进行空间细胞类型注释。RCTD拟合一个统计模型,该模型估计每个bin的细胞类型混合,文中的例子证明RCTD可以准确地发现模拟和真实空间转录组数据中细胞类型的定位。
RCTD可以将单个细胞类型或细胞类型混合物分配到HD的bins。它有三种模式:
1. Doublet mode:每个点分配1-2种细胞类型,推荐用于高空间分辨率的技术,如Slide-seq和MERFISH;
2. Full mode: 每个点分配任何数量的细胞类型,推荐用于空间分辨率较差的技术,如100微米分辨率Visium;
3. Multi mode: 双重模式的扩展,可以发现每个点两种以上的细胞类型,作为全模式的替代选择。这里我们选择Doublet模式,每个8um bin分配1-2种细胞类型。
RCTD分析输入文件:
Visium HD - Space Ranger outputs in 8µm bin size
filtered_feature_bc_matrix.h5
Single-cell data (Celltype annotated .rds)
在进行整合之前,我们需要在空间文件夹输出中将tissue_positions.parquet转换为tissue_positions.csv。目前,spacexr包无法识别tissue_positions.parquet。此外,spacexr正在空间文件夹中查找以tissue_positions开头的文件,这意味着如果tissue_positions.parquet和tissue_positions.csv都在空间文件夹中,则会混淆spacexr。因此,将parquet转换为CSV后,我们需要删除parquet文件或完全重命名parquet文件。
step1. 将空间坐标文件tissue_posistions.parquet转换成csv格式的tissue_positions.csv文件,并删除/spatial文件夹下tissue_posistions.parquet文件
library(arrow)
tissue_pos <- read_parquet("./SpaceRanger_HD_8micron/spatial/tissue_positions.parquet")
write.table(
tissue_pos,
"./SpaceRanger_HD_8micron/spatial/tissue_positions.csv",
col.names=TRUE,
row.names=FALSE,
quote=FALSE,
sep=","
)
system("rm ./SpaceRanger_HD_8micron/spatial/tissue_positions.parquet")
step2. 读取单细胞数据,构造reference;开始解卷积分析。
ImmuRef <- readRDS('./scRNA_Human_CRC_P1.rds')
CountRef <- GetAssayData(ImmuRef, slot = "counts")
CTRef<-ImmuRef@meta.data$Ident
CTRef<-gsub("/","_",CTRef)
CTRef<-as.factor(CTRef)
names(CTRef)<-rownames(ImmuRef@meta.data)
## 构造reference对象
reference <- Reference(CountRef[,names(CTRef)], CTRef , colSums(CountRef))
# Deconvolve HD Data
counts<-Read10X_h5("./SpaceRanger_HD_8micron/spatial/filtered_feature_bc_matrix.h5")
coords<-read_parquet("./SpaceRanger_HD_8micron/spatial/tissue_positions.parquet", as_data_frame = TRUE)
coords <- as.data.frame(coords)
rownames(coords)<-coords$barcode
coords<-coords[colnames(counts),]
coords<-coords[,3:4]
nUMI <- colSums(counts)
puck <- SpatialRNA(coords, counts, nUMI)
barcodes <- colnames(puck@counts)
myRCTD <- create.RCTD(puck, reference, max_cores = 20)
myRCTD <- run.RCTD(myRCTD, doublet_mode = 'doublet')
RCTD_result <- data.frame(ID=rownames(myRCTD@results$results_df), myRCTD@results$results_df)
write.table(RCTD_result, file = file.path(sample_dir, 'RCTD_result.txt'), sep = '\t', row.names = FALSE, col.names = TRUE)
step3. 解卷积结果展示
adata = sc.read_h5ad('./adata_P1.h5ad')
rctd_res = pd.read_csv('./RCTD_result.txt', index_col=0)
rctd_res = rctd_res.dropna()
adata.obs = pd.merge(adata.obs, rctd_res, left_index=True, right_index=True, how='left')
adata = adata[~adata.obs['first_type'].isna()]
with rc_context({'figure.figsize': (8, 8)}):
ov.pl.embedding(
adata,
basis='X_umap',
color=['first_type'],
size=4,
show=False,
title='RCTD CellType',
add_outline=False,
frameon='small',
legend_fontoutline=2,
)
with rc_context({'figure.figsize': (8, 8)}):
sc.pl.spatial(
adata,
color=['first_type'],
title='RCTD CellType',
library_id='P1',
alpha_img=0.3
)
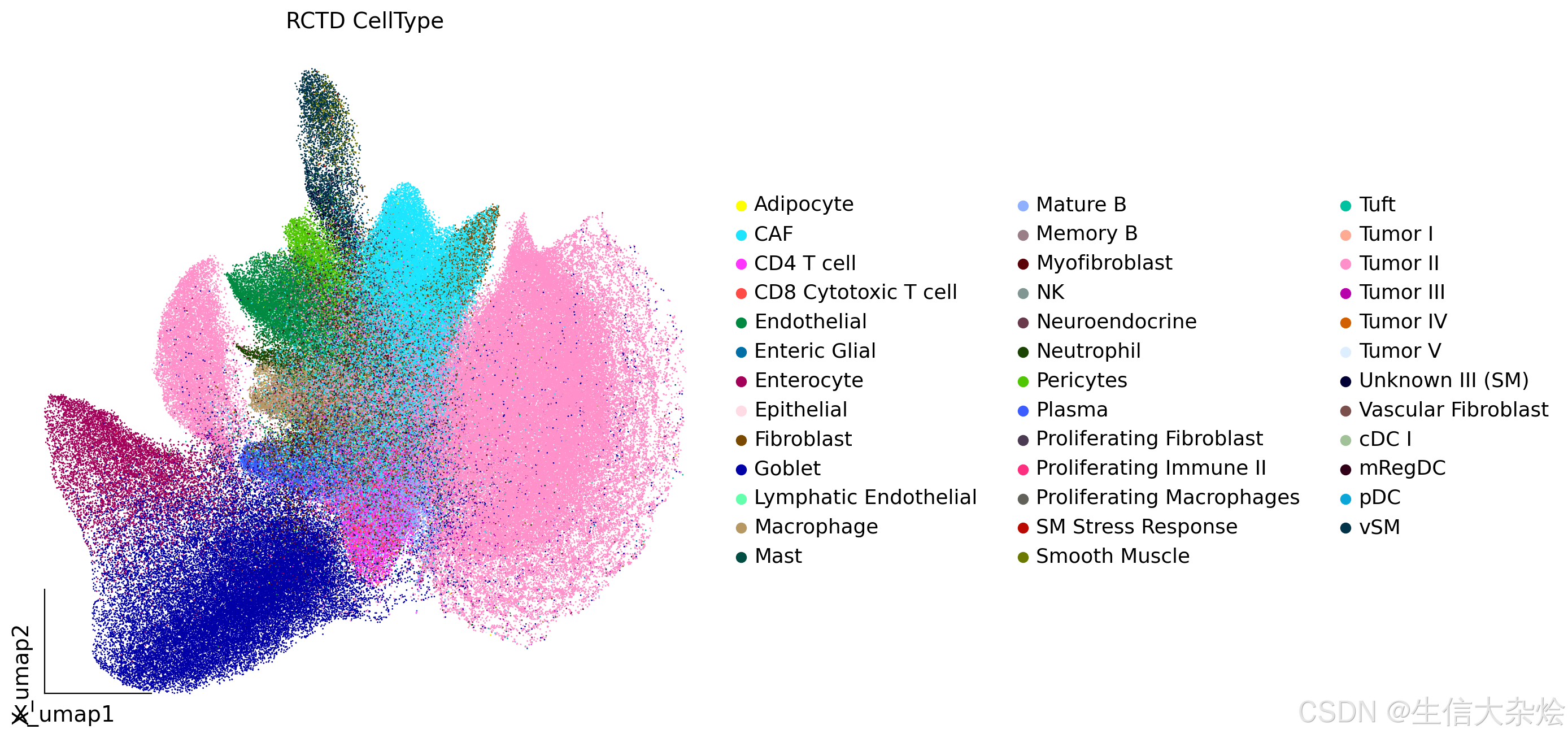
UMAP展示细胞类型注释结果
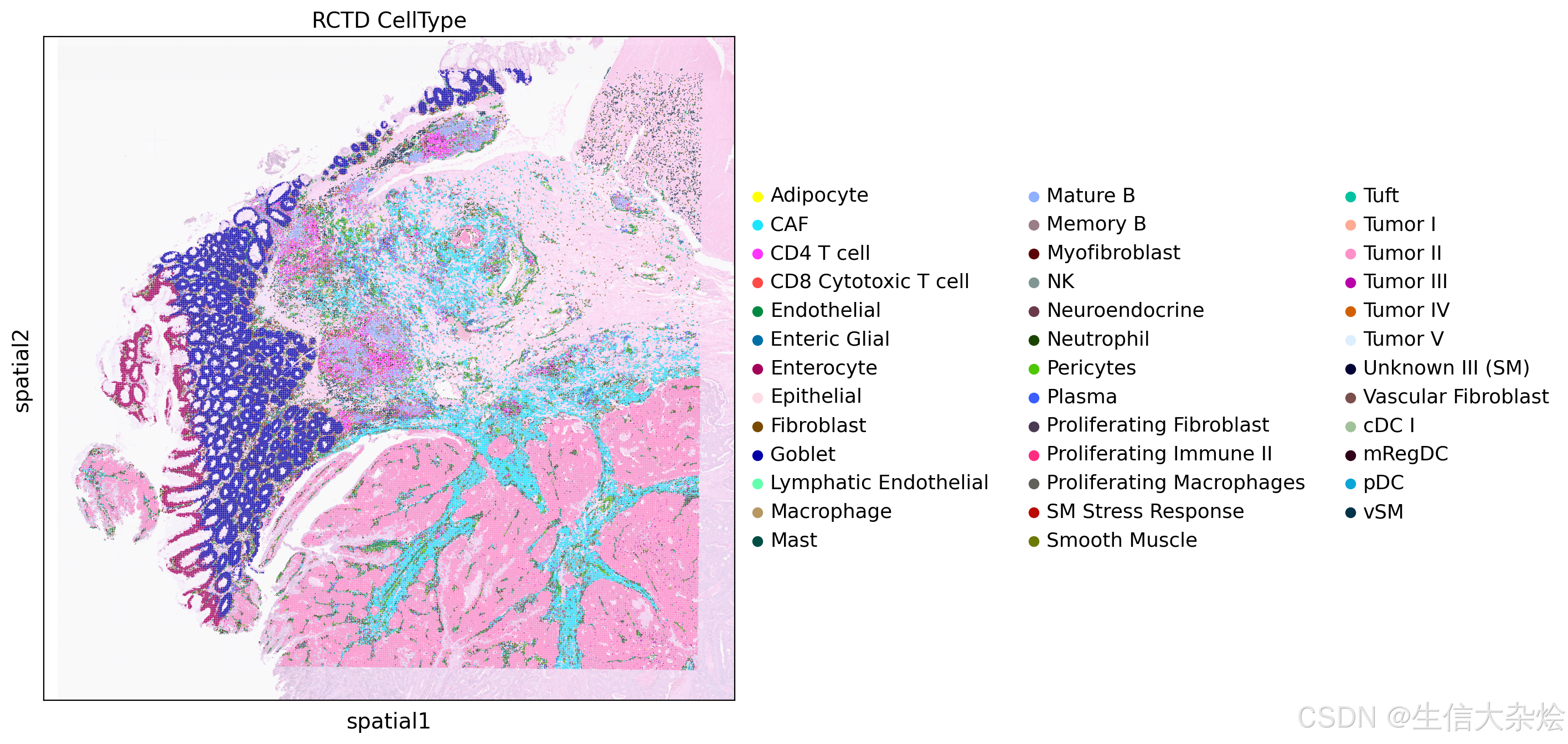
空间切片展示细胞类型
拿到单细胞注释结果后就可以进行一系列基于空间的后续分析了,包括邻域分析、空间邻近度分析、最近邻分析、细胞空间共定位、细胞空间通讯、空间信号流等个性化分析了。
微信公众号: 生信大杂烩


















 2万+
2万+

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









