







目录
ImmuCellAI-mouse平台的算法基于单样本基因集富集分析(single-sample Gene Set Enrichment Analysis,ssGSEA)。ssGSEA是一种常用的基因集富集分析方法,适用于单个样本的基因表达数据。
ImmuCellAI-mouse利用ssGSEA算法对癌症模型中的基因组数据进行分析。它将预定义的免疫相关基因集(例如与T细胞、B细胞、巨噬细胞等免疫细胞类型相关的基因)与样本中的基因表达数据进行比较。通过计算每个样本中免疫基因集的富集分数,可以得出不同免疫细胞类型在该样本中的相对丰度。
这种基于ssGSEA的分析方法能够帮助研究人员定量评估肿瘤组织中免疫细胞的浸润程度,提供关于肿瘤免疫环境的重要信息。通过深入了解不同免疫细胞类型的存在情况和丰度变化,研究人员可以得出更准确的结论,并为免疫治疗策略的设计和优化提供指导。
如果你有兴趣使用该平台进行分析,以下是一些步骤和资源供你参考:
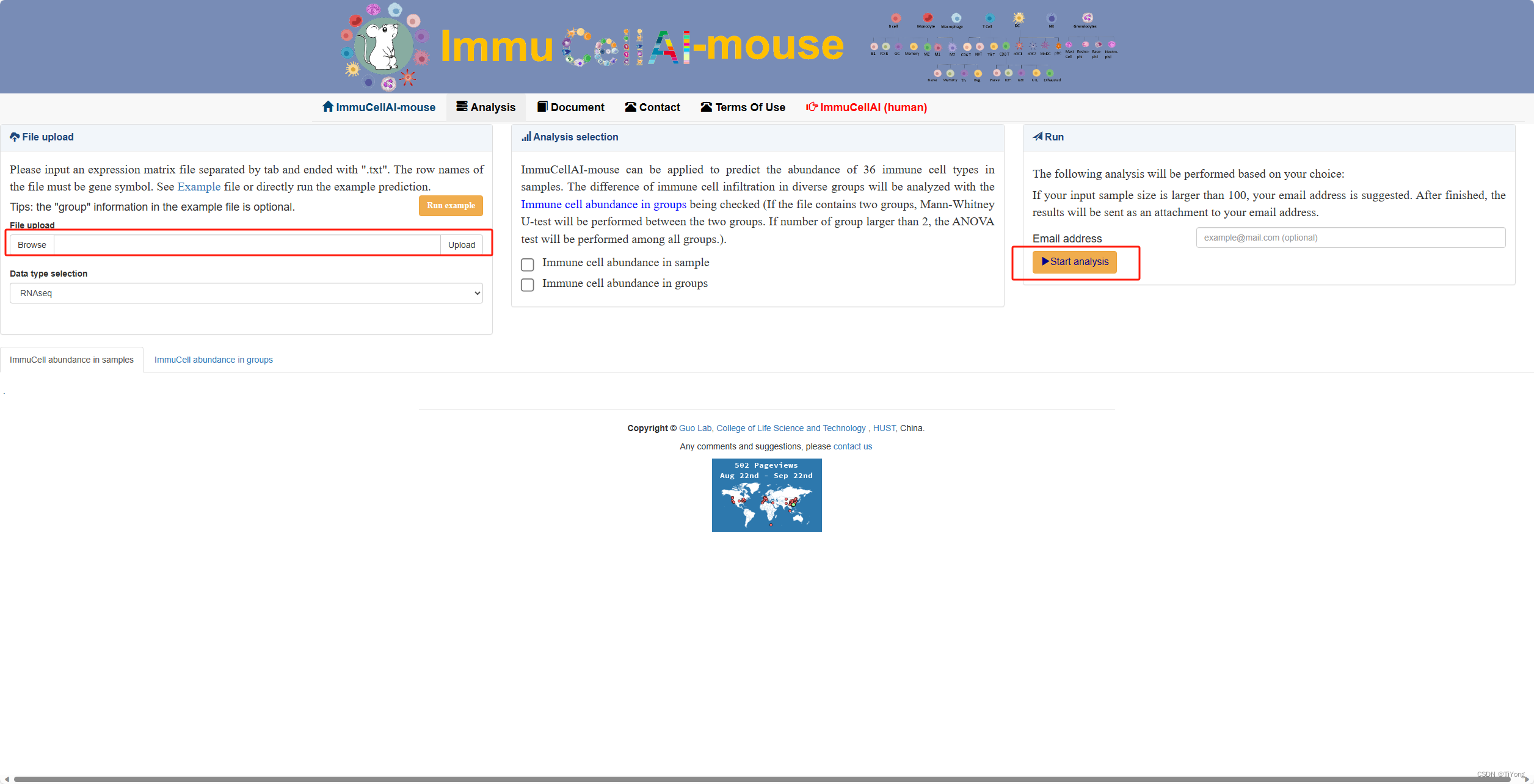
1. 在线工具:
在这里可以找到在线分析工具地址:
2. ImmuCellAI-mouse包的安装及使用
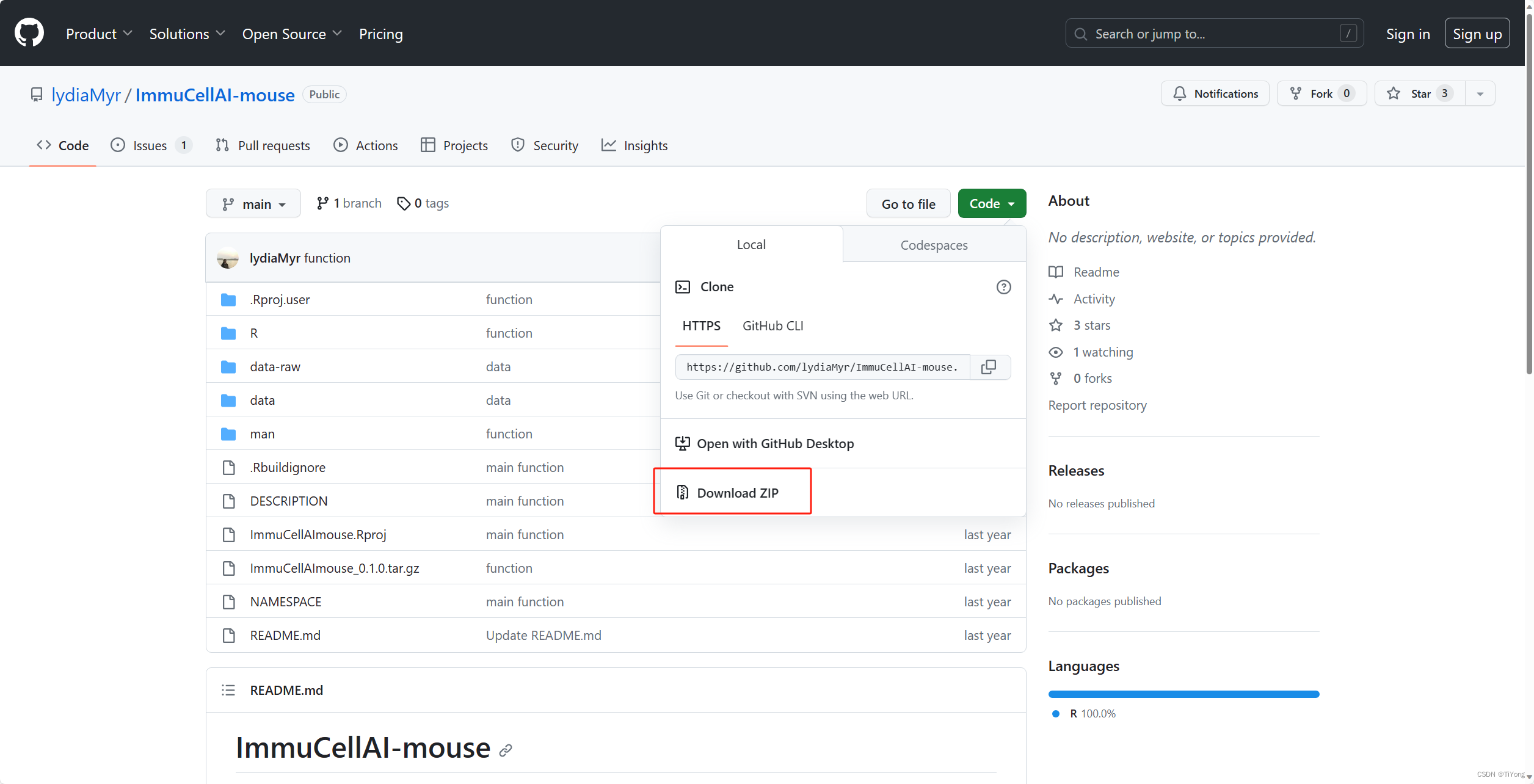
2.1 下载github包
通过以下链接下载ImmuCellAI-mouse的源代码:
https://github.com/lydiaMyr/ImmuCellAI-mouse/archive/refs/heads/main.zip
2.2 打开RStudio
打开RStudio,在接下来的步骤中会使用到它。
2.3 安装Rtools
需要先安装Rtools。你可以在这里下载Rtools的安装包:
https://mirrors.tuna.tsinghua.edu.cn/CRAN/bin/windows/Rtools/rtools43/files/
下载后,直接双击安装.exe文件。
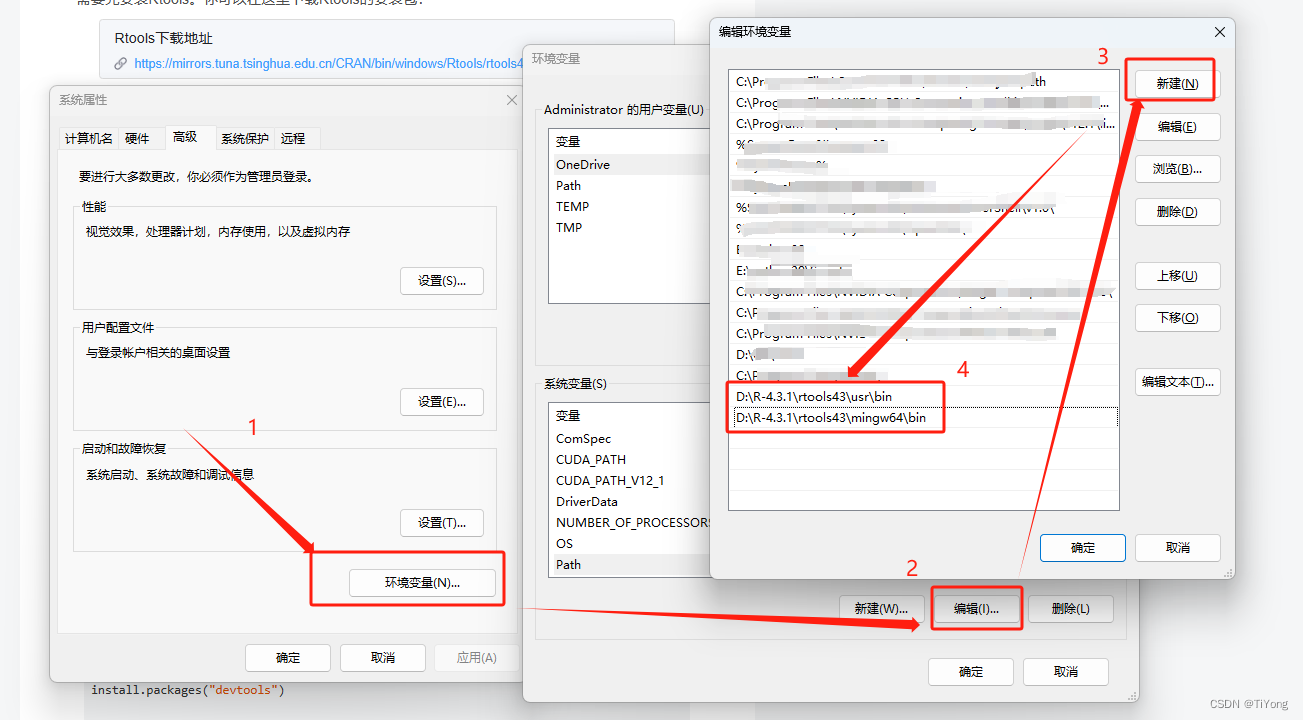
接下来,你需要将Rtools与RStudio进行绑定。
打开系统环境变量,添加以下两条变量(根据你自己的安装路径进行替换,例如,我的在D盘):
D:\R-4.3.1\rtools43\usr\bin
D:\R-4.3.1\rtools43\mingw64\bin
在RStudio的脚本或命令行中输入以下代码来检查是否将RTools与RStudio绑定:
install.packages("jsonlite", type = "source")
如果没有报错,则表示安装成功。如果报错,请自行搜索解决方法,确保成功安装了RTools,并继续下一步操作。
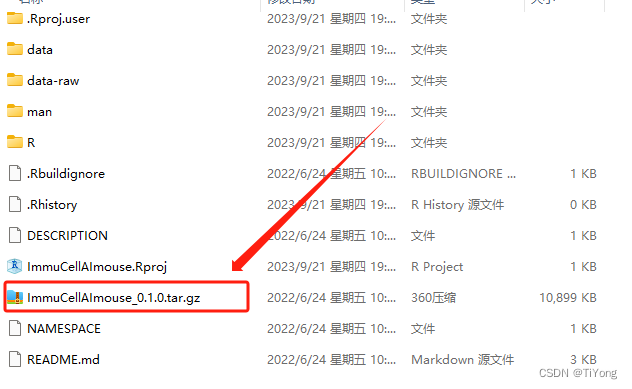
2.4 安装ImmuCellAI-mouse
压缩第一步下载好的包后,在RStudio中执行以下代码来安装:
install.packages("devtools")
library(devtools)# 安装解压后的文件,ImmuCellAImouse_0.1.0.tar.gz,输入文件地址:(替换为你解压后的地址)
# 安装解压后的文件,ImmuCellAImouse_0.1.0.tar.gz,输入文件地址:(替换为你解压后的地址)
install.packages("B:/ImmuCellAI-mouse-main/ImmuCellAImouse_0.1.0.tar.gz",
repos = NULL,
type = "source")# 上一步会需要许多相关的包,根据自己缺少的包进行安装。
# 安装完成后,加载包
library(ImmuCellAImouse)2.5 使用官方数据验证安装结果
接下来,你可以通过以下代码来查看该包提供的数据以及加载官方实验数据的示例:
看一下官方数据:这样:
# 查看包提供的数据
data(package="ImmuCellAImouse")
# 加载官方实验数据:
data(ImmuCellAI_mouse_example,package = "ImmuCellAImouse")
# 将第一列设置为行名
rownames(ImmuCellAI_mouse_example) <- ImmuCellAI_mouse_example[,1]
ImmuCellAI_mouse_example <- ImmuCellAI_mouse_example[,-1]
# 查看数据
head(ImmuCellAI_mouse_example)
# 运行分析函数
test <- ImmuCellAI_mouse(sample =ImmuCellAI_mouse_example,
data_type = "rnaseq",#数据类型,可选"rnaseq"/"microarray",即你输入的数据类型
group_tag = 1,#是否有分组信息,如果没有则填"0"
customer=FALSE)# 是否有自行上传的参考文件,有"1"无"0",一般来说不用上传
#查看结果
names(test)#输出了两个结果,"abundance"是丰度结果,
# "group_result"是添加了分组信息的结果,如果 group_tag = 0,则该list为NULL
#丰度信息
head(test$abundance)
#分组信息
head(test$group_result)3. 测试自己的代码
如果你有自己的数据,只需将sample参数改为你的数据即可。
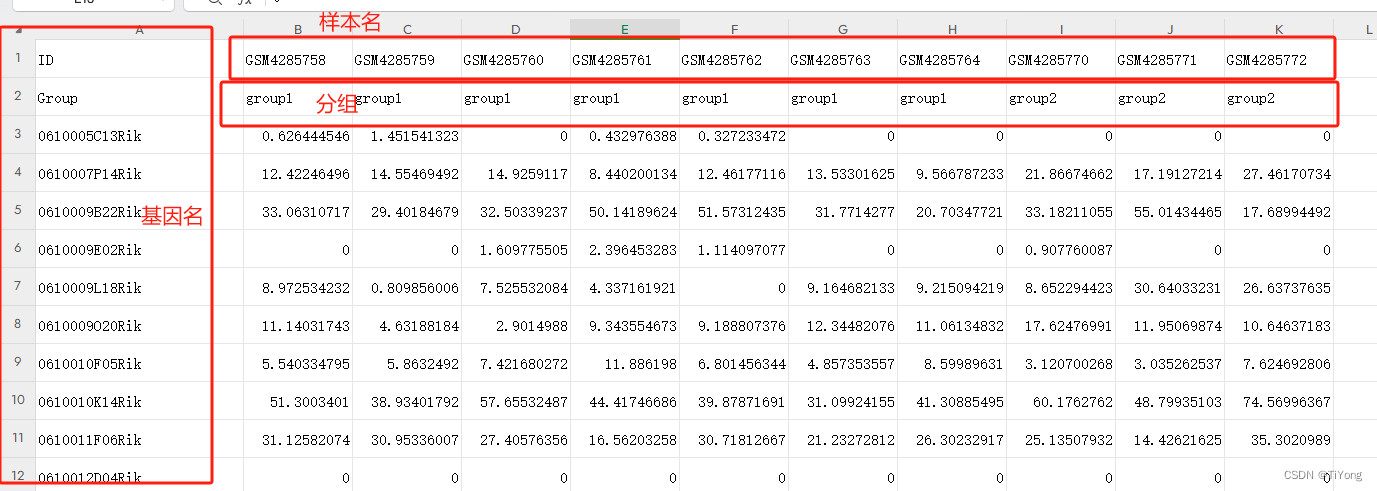
用测试数据讲一下,这里有三个细节
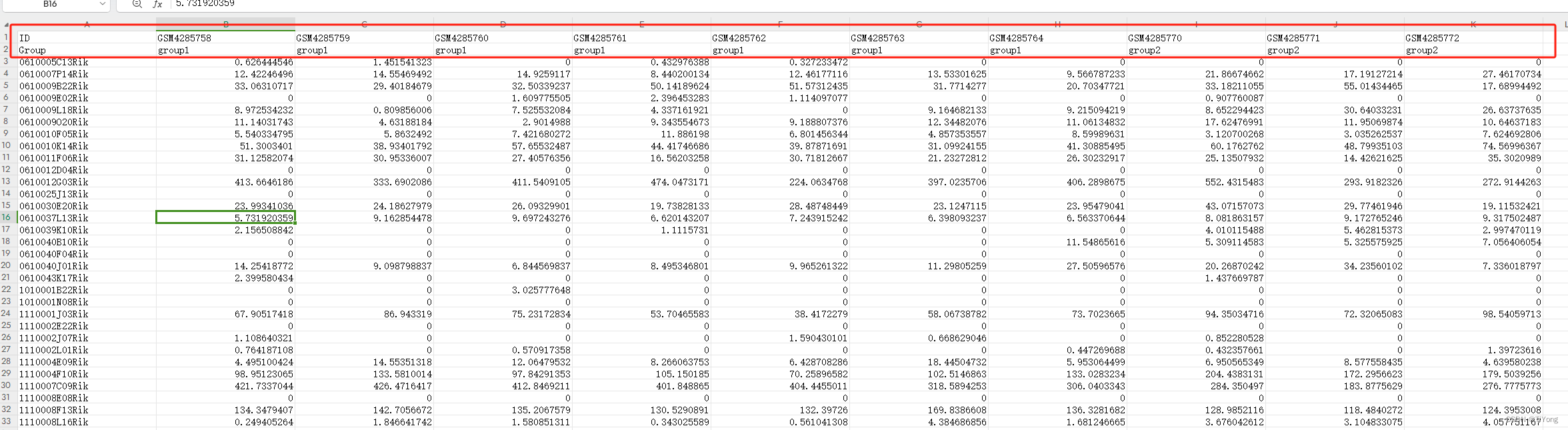
1.第一行为 ID 样本名
2.第二行为 Group 分组信息
3.第一列为基因名。
4.数据为TPM值
例如:
data <- read.table("B:/ImmuCellAI-mouse/sample.txt", header = TRUE, sep = "\t", row.names = 1)
test <- ImmuCellAI_mouse(sample =data,
data_type = "rnaseq",#数据类型,可选"rnaseq"/"microarray",即你输入的数据类型
group_tag = 1,#是否有分组信息,如果没有则填"0"
customer=FALSE)# 是否有自行上传的参考文件,有"1"无"0",一般来说不用上传
最后提一句:数据的行,也就是基因数,建议超过5000行,而且建议是symbol ID。如果数据过过少,会出现维度不匹配报错,原因是因为会在会在免疫细胞分层的过程中,合并数据,如果基因过少,合并的维度就不一致,会报错。(这里不知道对不对,需要大佬指教一下。)
如果你想深入学习有关ImmuCellAI-mouse的更多内容,
推荐阅读下面这篇文章:ImmuCellAI-mouse R包学习指南。
博主写得很好:Aech_-优快云博客
希望通过ImmuCellAI-mouse平台能够帮助你在癌症基因组大数据分析方面取得更好的结果!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











