







在生物信息学分析中,从数据库(如 NCBI 或 ENA)下载原始测序数据(如 FASTQ 文件)是常见的第一步。然而,传统方式通常需要手动查询 SRA 号、安装复杂的工具、甚至编写冗长的脚本,这对入门用户和追求效率的研究者来说,都显得繁琐耗时。
这篇文章为大家介绍一款高效、简洁的生信下载工具——iSeq。通过 iSeq,我们可以实现用“一行代码”快速下载原始测序数据,极大地提升工作效率!本文将重点介绍 iSeq 的功能特点及使用方法,帮助你轻松上手。
一、简介
iSeq是一个Bash脚本工具,可以方便快捷从GSA、SRA、ENA和DDBJ数据库下载测序数据和元数据。
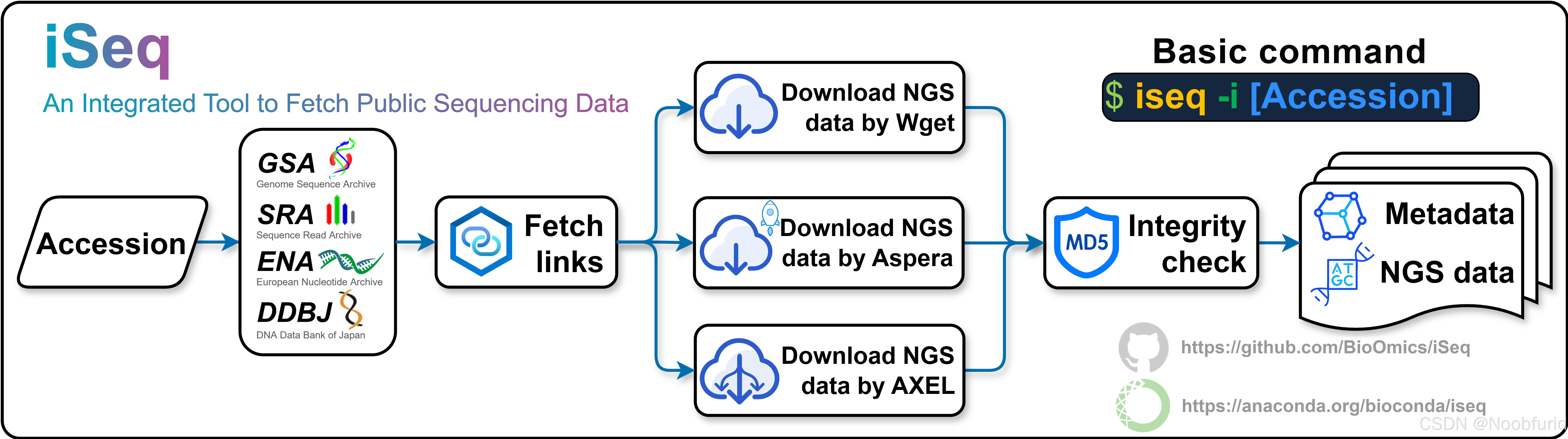
项目GitHub地址:https://github.com/BioOmics/iSeq
项目引用: Haoyu Chao, Zhuojin Li, Dijun Chen, Ming Chen, iSeq: An integrated tool to fetch public sequencing data, Bioinformatics, 2024;, btae641, iSeq: an integrated tool to fetch public sequencing data | Bioinformatics | Oxford Academic
二、使用方法
1. 安装iSeq
首先系统上需要安装有conda环境(下载Conda:Download Now | Anaconda),在安装iSeq之前可以先创建一个专门用来下载的虚拟环境:
# 创建conda虚拟环境
conda create -n iseq虚拟环境创建好之后启动该环境:
# 启动虚拟环境
conda activate iseq在虚拟环境中安装iSeq:
# 安装iSeq
conda install bioconda::iseq安装完成后查看iSeq版本:
iseq --version2. 使用iSeq下载测序数据
iSeq支持下载以下系列的数据,基本上覆盖了绝大多数的数据库:
| Databases | BioProject | Study | BioSample | Sample | Experiment | Run |
|---|---|---|---|---|---|---|
| GSA | PRJC | CRA | SAMC | \ | CRX | CRR |
| SRA | PRJNA | SRP | SAMN | SRS | SRX | SRR |
| ENA | PRJEB | ERP | SAME | ERS | ERX | ERR |
| DDBJ | PRJDB | DRP | SAMD | DRS | DRX | DRR |
| GEO | GSE | \ | GSM | \ | \ |
下面是这些数据库的一些示例:
| Accession Type | Prefixes | Example |
|---|---|---|
| BioProject | PRJEB, PRJNA, PRJDB, PRJC, GSE | PRJEB42779, PRJNA480016, PRJDB14838, PRJCA000613, GSE122139 |
| Study | ERP, DRP, SRP, CRA | ERP126685, DRP009283, SRP158268, CRA000553 |
| BioSample | SAMD, SAME, SAMN, SAMC | SAMD00258402, SAMEA7997453, SAMN06479985, SAMC017083 |
| Sample | ERS, DRS, SRS, GSM | ERS5684710, DRS259711, SRS2024210, GSM7417667 |
| Experiment | ERX, DRX, SRX, CRX | ERX5050800, DRX406443, SRX4563689, CRX020217 |
| Run | ERR, DRR, SRR, CRR | ERR5260405, DRR421224, SRR7706354, CRR311377 |
下载相应数据只需要一行代码:
# 下载数据
iseq -i accession
# accession替换成你想要下载的数据集的编号,例如:
# iseq -i PRJNA211801
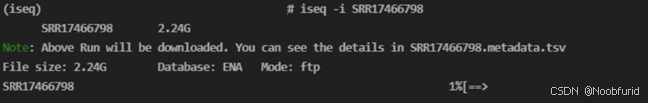
# iseq -i CRR343031程序会自动开始下载(需要支持FTP、HTTP 和 HTTPS 协议):
3. iSeq 一些其他参数
-m, --metadata
只下载accession的样本信息,跳过测序数据的下载。
iseq -i PRJNA211801 -m
iseq -i CRR343031 -m-g, --gzip
直接下载gzip格式的FASTQ文件,如果不能直接下载,则会下载SRA文件并通过多线程分解和压缩转换为gzip格式。
iseq -i SRR1178105 -g-q, --fastq
将下载完成的SRA文件分解为多个未压缩的FASTQ格式。
iseq -i SRR1178105 -q-t, --threads
指定分解SRA文件为FASTQ文件或者压缩FASTQ文件的线程数,默认为8。
iseq -i SRR1178105 -q -t 10-e, --merge
将Experiment中的多个FASTQ文件合并为一个FASTQ文件
iseq -i SRX003906 -g -e ex-d, --database
指定下载SRA文件的数据库,支持ena和sra两种数据库。
iseq -i SRR1178105 -d sra -p, --parallel
开启多线程下载,需要指定下载的线程数。
iseq -i PRJNA211801 -p 10-a, --aspera
使用Aspera进行下载。
iseq -i PRJNA211801 -a -g-o, --output
v1.1.0之后可以选择输出文件的位置,如果不存在相应的文件夹,则创建
iseq -i SRR931847 -o PRJNA211801-s, --speed
v1.2.0之后可以选择限速下载,单位是MB/s
iseq -i SRR931847 -s 10其他详细参数的解释可以到项目地址进行阅读:iSeq/docs/ChineseTutorial.md at main · BioOmics/iSeq · GitHub


















 2096
2096

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











