







1.相关资料
1.1.推荐文章
Justin指引:英文版(必看!)
http://www.mdtutorials.com/gmx/complex/index.html
来源知乎:《GROMACS分子动力学模拟》
https://zhuanlan.zhihu.com/p/160899845
来源知乎:《速通Windows版gromacs安装(gromacs+小分子准备+mmpbsa计算+分析图展示)》
https://zhuanlan.zhihu.com/p/678585456
来源优快云:《Gromacs基础教程一:入门建议》
https://blog.youkuaiyun.com/m0_67843839/article/details/127537660
来源优快云:《水中的溶菌酶 第一步:准备拓扑》
https://blog.youkuaiyun.com/m0_67843839/article/details/127895765
思想公社相关专栏:
http://sobereva.com/category/Modelling/1/
1.2.其他文章
Jerkwin中文版Gromacs教程:已经过时了
`https://jerkwin.github.io/9999/10/31/GROMACS%E4%B8%AD%E6%96%87%E6%95%99%E7%A8%8B/
1.3.推荐视频
来源B站,强推。如果说1.3中只能推荐一个视频的话,建议看这个。哪怕你要做的是蛋白-配体复合物体系,也应该先过一遍这个。
来源B站,讲的都是有效的概论,其中关于分子动力学模拟归纳的三步挺有用的。

来源B站,没有讲关于分子对接后得到的文件该如何处理。但是还是推荐看下的

来源B站,需要用的是autodock vina,而且走过一遍up主之前分子对接的视频才比较好理解。很容易卡在complex.pdb里面都是ATOM开头的行这一步。

来源B站,纯操作视频,没有废话。不过没有前置讲解,譬如分子对接后的pdb格式文件怎么来的,没有讲解操作背后的含义,以及看不到鼠标,画质堪忧hhh

来源youtube,如果你能忍受印度老哥的口音的话,讲的还行。
1.4.其他视频
来源B站,讲的太过概论了,不适合新手上手。

来源B站,蕴含的信息太少。

来源B站,说实话并不知道为什么播放量这么高,其实没有讲很多东西啊hhh

来源B站,这是一个用很小众的YASARA软件去做分子对接和分子动力学模拟的教程,up主全程没有废话,讲的很好。不过貌似现在这个软件能下载到的版本和视频中的有些许出入,同时并不知道是开源与否,论文是否能引用。up主评论区给的回复是可以自己做实验时用这个软件,写论文时引用别的软件或网站hhh。好处就是该软件不像mgltools和autodockvina一样吃电脑配置,cpu为4核也能跑。坏处就是它有自己的格式文件,如sce和yob,不是用的常用的pdbqt格式。

2.分子动力学模拟(MD)
2.1.什么是分子动力学模拟(MD)
在分子动力学模拟(MD)中,分子体系内的原子被赋予初始位置和速度,然后根据它们之间的相互作用力(这些力可以基于经验势函数或更复杂的量子力学计算),计算出每个原子的加速度。通过数值积分方法,逐步推进时间,可以计算出原子在下一时刻的位置和速度,这样就可以模拟出整个体系随时间的演化过程。
2.2.MD研究的方向和局限性
MD研究的问题,最经典的就是酶和底物的结合问题,譬如大分子蛋白和小分子配体的结合。其结合问题又可以细分为:
- 蛋白上哪里是结合位点?
- 哪些小分子可以结合上去?
- 结合自由能是多少?
- ……等等
同时,MD也用来研究酶的动态过程,譬如钾离子通道通过MD后发现钾离子是一个挨着一个通过的,而不是钾离子间隔着水排列通过的,这颠覆了人们的最初的猜想,毕竟钾离子带着正电,人们以为它们不会间隔很近。
但MD也有一定的局限性,譬如我们需要考虑算力的大小,当一个模拟的蛋白过大(譬如为200A时),或者结合时间过短,模拟的难度就会很高。
同样的,因为MD是以力场来描述整个体系的,如果一个结合过程涉及到除了经典力学之外的很多其他反应,譬如化学键的成断,这也往往是用其他工具去研究的,譬如QM/MM。
2.3.MD的一般步骤
一个分子动力学模拟的一般步骤如下:
- 创建系统初始状态
- 生成蛋白的拓扑文件
- 添加盒子和系统的溶剂化作用
- 添加离子使得整个系统达到平衡
- 引入系统的相互作用势能
- 能量最小化(目的是为了让整个系统更加稳定)
- 选择系综(或者说选择系统条件,譬如是等温等压还是等压等焓)
- 预测粒子如何移动
- 平衡模拟
2.3.MD的结果分析
对于蛋白-配体复合物体系,我们结果往往关注:RMSD、RMSF、氢键、Rg、自由能、DSSP、最稳定构象簇。
(1)RMSD可以观察到小分子的位置变化,我们希望小分子的曲线一直小于0.2m.
(2)RMSF和DSSP图则可以观察到哪些序号的氨基酸残基的构象变化比较大。
(3)氢键图有利于观察小分子和蛋白质之间形成的相互作用力情况。
(4)复合物的自由能的计算方法可以使用MMPBSA,出自Jerkwin老师的脚本gmx mmpbsa.bsh文件。
(5)自由能形貌图则可以得知本次模拟过程中形成的最低能量构象。
2.4.MD中相关注意点
2.4.1.生成小分子拓扑文件
gromacs并不可以生成小分子拓扑文件,现在常见的小分子拓扑文件生成工具有:
charmm series、OPLS-AA、sobtop、AMBERTOOLS、MMFF series、SWISSPARAM。
小分子拓扑完美融入复合物体系的标志是可以生成含有水溶液的pdb文件。
注意,需要区分这些工具生成的小分子拓扑文件是纯拓扑文件还是带了位置限制文件的拓扑文件。
2.4.2.MD中小分子处理
在经历分子对接后,小分子往往被限制在了蛋白质的某个口袋区域里。
但是在分子动力学模拟时,我们需要有NVT温度平衡和NPT压力平衡的操作,这往往会导致小分子的位置偏移。为此我们需要额外注意小分子的位置限制文件。
- 如果分子对接的结果较为可观,比如最佳对接姿态里的结合能绝对值较大,就不用更改默认的限制参数。
- 如果分子对接的结合能绝对值比较低、受体配体间作用力比较少的情况下,建议大家可以先在NVT、NPT过程中保持默认参数,然后运行大概1ns或更久的MD作为测试标准),结束后观察小分子是否发生较大幅度的位置变化。再根据变化情况酌情调整限制参数。
如果是依赖非groamcs.工具生成的限制参数文件,那么可以使用notepad,打开列编辑模式进行手动调整。
2.5.MD算力要求
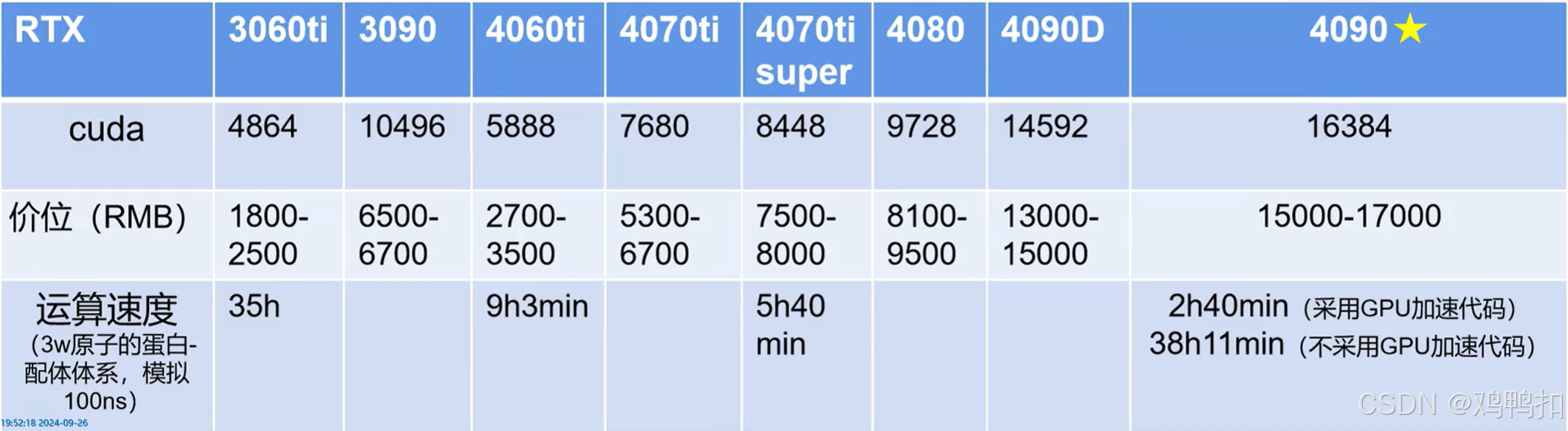
图源:B站up主DS医学生
时间:2024年7月京东
推荐超算平台:北京北龙超级云计算中心、成都天玑智算云平台。
3.gromacs
3.1.gromacs概述
gromacs是一款用来分子动力学模拟(Molecular Dynamics, 简称MD)的软件。
利用gromacs可以模拟到3kcal/mol的程度,虽然距离理想实验结论1kcal/mol仍有一定距离,但已经是个不错的数字了。

Justin版指引如上,教程1是水中的溶酶体。
3.2.原子类型
原子类型是分子力学中一个很重要的概念,计算的基础。
原子类型包括元素类型,原子杂化态,原子环境。
以碳原子为例,CH4中的碳与C2H4的碳是不同的,一个是SP3杂化,一个是SP2杂化。
3.3.力场分类与适用范围与推荐力场
力场可分为以下三种:
- 全原子力场:精确定义每一个原子的参数。
- 联合原子力场:省略非极性氢原子,同时把其参数整合到与他们成键的相邻原子上(比如甲基,只由一个碳原子表示,实际上和全原子力场相差不大。
- 粗颗粒力场:进一步精简分子结构的力场参数,种类比较多,比如有将蛋白侧链看作一个颗粒的力场,或者甚至将整个氨基酸残基看成一个颗粒的力场等等。
不同力场的适用范围:
- AMBER力场:目前使用比较广泛的一种力场,适合处理蛋白质、核酸、多糖等。为全原子力场;
- CHARMM力场:引用了大量的量子计算结果为依据。全原子力场,比较适合蛋白配体复合物体系。
- MMX力场:适用于各种有机化合物,自由基,离子。考虑了许多交叉作用项。
- GROMOS力场:联合原子力场
- OPLS-AA/L力场:适合溶液体系。
推荐使用力场:charmm36、ff19sb、gaff2,这三个是当前领域中最新、预测精度高的力场。ff19sb在蛋白质突变等方面具有更好的预测能力。
这三个力场都可以向下兼容到ff14sb、ff99sb、gaff、charmm27、gromos、OPLS-AA、UFF、MMFF94。
3.4.水模型
TIP3P/SPC/SPCE模型,有三个参数表示一个水分子,分别是O-H键长、O-O夹角角度、分子半径。

如果想模拟的是结晶的水模型,譬如冰晶等,用TIP4P/TIP5P模型。
4.MOL2、PDB格式解读
4.1.MOL2格式解读
用记事本打开一个MOL2文件,如下:
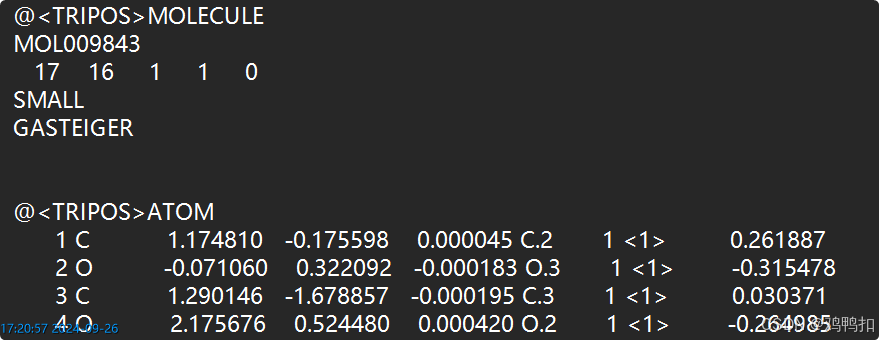
可以看到第二行的第一个数字17,就是指该分子有17个原子。
再往下,ATOM以下的部分,就是关于各个原子的详细描述。以第一行举例如下:
第二列C等为元素名。
第三列1.174810到第五列0.000045就是该原子在x y z上的位置。
第六列C.2即原子类型,因为不同的原子类型会有不同的电荷,譬如α-C和羰基碳虽然都是C元素,但是有着不同的电荷。再譬如这里第二行的O和第四行的O,一个是O.3,一个是O.2。
最后一列0.261887是该原子的电荷。
4.2.PDB格式解读
用记事本打开一个PDB格式文件,如下:

在这里我们先关注ATOM行。
第三列是原子类型,譬如N就代表是蛋白质的氨基端的氮原子,CA代表α-C(阿尔达碳原子),C代表羰基碳。
第四列,如THR,是氨基酸名称缩写。
第五列,如A,是指当前原子属于蛋白质的哪条链。
第六列,如412,是指当前原子属于第412位氨基酸残基的。
第七列到第九列,如11.133到26.540,是指当前原子的x y z位置参数。
第十列,如1.00,是指当前原子在前面位置参数出现的概率,这里为1,代表该原子在前面位置参数出现的概率为百分百,也就是变相说明了该原子的固定位置为前面位置参数。
第十一列,如54.12,是指当前原子的热震动程度。
第十二列,如N,是指元素名。
可以看到第十一列和第十二列中间空了一列,如果我们计算了该蛋白的电荷量,到时候再打开该PDB文件,那么该列会补充上每个原子的电荷量。



















 1790
1790

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











