






 本文介绍了化学信息学工具RDKit中用于计算分子三维性质的Plane of Best Fit (PBF)方法,详细阐述了加氢、生成3D构象及计算PBF的步骤。PBF依赖于分子的3D坐标,因此在计算中需要固定randomSeed以确保一致性。博主分享了自己的代码实现,并指出对于比较数据库中的分子,使用相同randomSeed的重要性。
本文介绍了化学信息学工具RDKit中用于计算分子三维性质的Plane of Best Fit (PBF)方法,详细阐述了加氢、生成3D构象及计算PBF的步骤。PBF依赖于分子的3D坐标,因此在计算中需要固定randomSeed以确保一致性。博主分享了自己的代码实现,并指出对于比较数据库中的分子,使用相同randomSeed的重要性。
写在前面: 本人是一个药学研究生小白,这是第一次将平时遇到的以及学习的部分知识进行记录整理,如若觉得写的内容不通顺等还希望能理解,如果感觉有错误欢迎批评指正,谢谢!
关于RDKit
RDKit 是开源的化学信息学python软件包,功能特别强大。通过RDkit这个工具包基本可以实现很多对分子的二维以及三维性质的计算,各类fingerprint指纹生成,化合物结构相似性计算等等。
关于PBF
今天要讲述的是化合物分子其中一个三维性质的计算方法——PBF(Plane of Best Fit)。这是我在研究生学习以及动手实验过程中遇到的一个需要计算的性质。该性质论文具体参见以下链接。Plane of Best Fit: A Novel Method to Characterize the Three-Dimensionality of Molecules | Journal of Chemical Information and Modeling (acs.org) https://pubs.acs.org/doi/10.1021/ci300293f
https://pubs.acs.org/doi/10.1021/ci300293f
简要来说,PBF(Plane of Best Fit)——最佳拟合平面,是作者用来量化和表征分子的一个新的三维特征。该计算方法计算快速,易于分析一些大型的数据集。该方法将分子去除盐,然后用CORINA生成坐标。利用所得坐标计算出最佳拟合平面。 用最小二乘法求解最佳拟合平面。
使用RDkit进行PBF性质计算
在RDkit中使用以下方法进行PBF计算,可在RDkit官方文档中进行查找。
rdkit.Chem.rdMolDescriptors.CalcPBF((Mol)mol[, (int)confId=-1])
Returns the PBF (plane of best fit) descriptor
C++ signature :
double CalcPBF(RDKit::ROMol [,int=-1])但RDkit对如何使用该方法的具体细节并没有进行讲解。这里我讲述下我在使用该方法时的流程,并贴上我自己写的脚本中的代码,尝试过是可以成功计算的。
1.加氢
加氢: RDkit中默认不显示氢,因为是三维性质计算,所以对其进行加氢得到的结果会比不加氢结果要好。以下方法执行分子加氢计算。
Chem.AddHs(mol)2.生成3D构象
使用距离几何算法初始化3D坐标。
AllChem.EmbedMolecule()((Mol)mol[, (int)maxAttempts=0[, (int)randomSeed=-1[, (bool)clearConfs=True[, (bool)useRandomCoords=False[, (float)boxSizeMult=2.0[, (bool)randNegEig=True[, (int)numZeroFail=1[, (dict)coordMap={}[, (float)forceTol=0.001[, (bool)ignoreSmoothingFailures=False[, (bool)enforceChirality=True[, (bool)useExpTorsionAnglePrefs=True[, (bool)useBasicKnowledge=True[, (bool)printExpTorsionAngles=False[, (bool)useSmallRingTorsions=False[, (bool)useMacrocycleTorsions=False[, (int)ETversion=1]]]]]]]]]]]]]]]]]) → int rdkit.Chem.rdDistGeom.EmbedMolecule((Mol)mol[, (int)maxAttempts=0[, (int)randomSeed=-1[, (bool)clearConfs=True[, (bool)useRandomCoords=False[, (float)boxSizeMult=2.0[, (bool)randNegEig=True[, (int)numZeroFail=1[, (dict)coordMap={}[, (float)forceTol=0.001[, (bool)ignoreSmoothingFailures=False[, (bool)enforceChirality=True[, (bool)useExpTorsionAnglePrefs=True[, (bool)useBasicKnowledge=True[, (bool)printExpTorsionAngles=False[, (bool)useSmallRingTorsions=False[, (bool)useMacrocycleTorsions=False[, (int)ETversion=1]]]]]]]]]]]]]]]]]) → int :
在继续PBF性质计算之前,要注意EmbedMolecule的一个参数randomSeed。计算PBF的时候同一个分子几次计算结果不同的原因就在这。
PBF是一种三维属性,它是特定于三维构象的,依赖于三维坐标。
每次调用EmbedMolecule()都会生成一个以不同的3D坐标集为特征的构象,可以通过调用molh.GetConformer(). getpositions()来检查。因此,每次PBF计算结果的不同取决于每次EmbedMolecule()生成的3D坐标集。
只需要将randomSeed参数设置为固定整数,这样每次调用EmbedMolecule()都会得到相同的3D坐标集。再进行PBF计算因此会得到相同的PBF值。
同时要注意查看AllChem.EmbedMolecule()的返回值。 它返回添加到分子中的构象的ID,如果嵌入失败则返回 -1 。
3.计算PBF性质
以上步骤均正常之后便可进行如下PBF计算了。
Chem.rdMolDescriptors.CalcPBF(mol)以下附上我的代码。
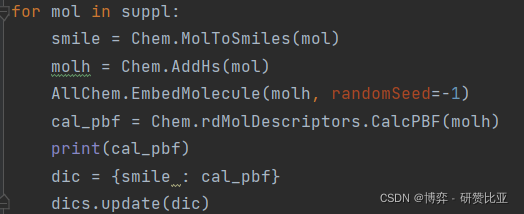
针对randomSeed参数并没有说具体哪个数值更好。不过用于比较数据库的话,用固定的同一个数值计算数据库中所有分子应该没有太大问题。
写在后面: 第一次分享,鉴于小白程度可能理解的没有很深入,欢迎大家探讨以及批评指正。或者有人知道某类分子有某个randomSeed最佳值也希望能告诉我做个补充。谢谢!



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言











