






生信碱移
GESECA分析
GESECA 是一种筛选具有高基因相关性的基因集的方法,能够在没有分组信息的情况下筛选重要的通路,并且能在bulk、单细胞以及空间转录组数据中进行应用。
单细胞与空间多组学让我们能够在细胞/空间分辨率的情况下观察病理或生理相关的转录变化。基因集是一组具有类似功能或相同途径的基因集集合,在多组学分析中,常常观察特定基因集的表达变化来推断相应功能途径的活性。举个例子,在一些肾炎相关疾病中,铁死亡基因集发生显著表达变化影响疾病进展。
常规的分析流程基于分组数据进行比较,比如 GSEA 使用 logFC 排序的基因列表观察特定基因集的表达变化。尽管如此,大多数据中通常没有明显的分组信息用于差异分析。一般来说,同一个基因集内部的基因变化是密切相关的。GESECA 是基于这个假设的基因集共调控分析方法,能够在没有分组信息的情况下筛选重要的通路,并且能在bulk、单细胞以及空间转录组数据中进行应用。
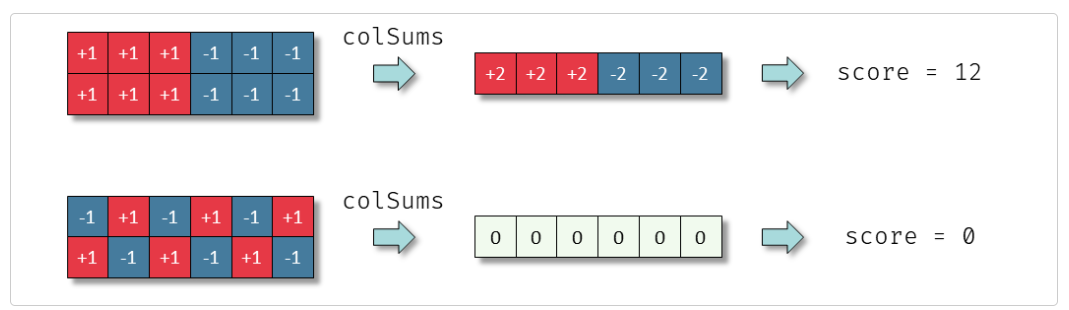
▲ GESECA 原理示意:GESECA 通过分析基因表达矩阵,计算每个基因集合在数据中的方差贡献,以衡量其在样本中的整体影响。如上图所示,具有一致波动变化的基因集会具有更高的得分。其核心思想借鉴了主成分分析(PCA),将基因集合的得分视为该集合解释的方差信息。高度相关的基因在得分中会相互增强,而不相关的基因则会相互抵消,从而突出真正具有生物学意义的基因集合。相比传统方法,GESECA 不依赖于样本注释或分组对比,使其适用于多种测序技术(如 RNA-seq、单细胞测序、空间转录组)。
GESECA 分析可以通过 fgsea 调用,该工具github链接如下:
-
https://github.com/alserglab/fgsea
0.R包安装
通过以下R包进行安装:
library(devtools)
install_github("ctlab/fgsea")
1.单细胞中应用
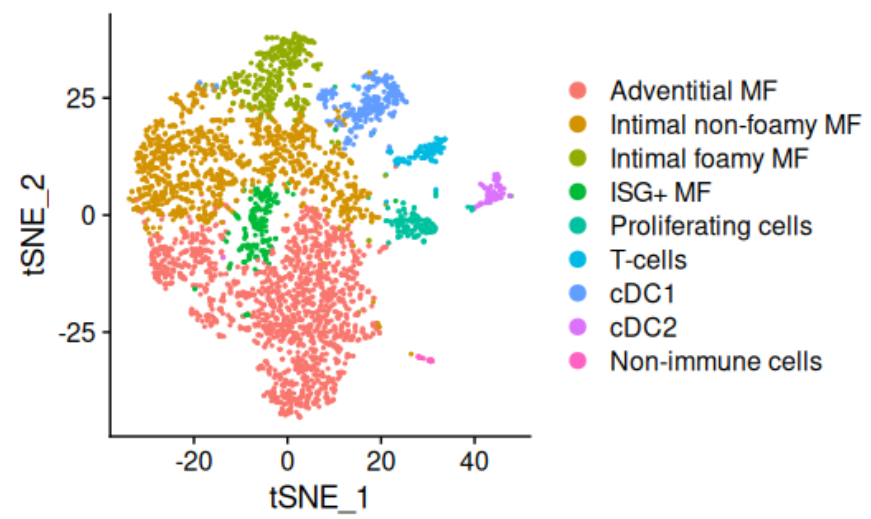
① 示例数据读入与单细胞人工注释:
library(Seurat)
# 读取seurat对象
obj <- readRDS(url("https://alserglab.wustl.edu/files/fgsea/GSE116240.rds"))
# 注释细胞与展示
newIds <- c("0"="Adventitial MF",
"3"="Adventitial MF",
"5"="Adventitial MF",
"1"="Intimal non-foamy MF",
"2"="Intimal non-foamy MF",
"4"="Intimal foamy MF",
"7"="ISG+ MF",
"8"="Proliferating cells",
"9"="T-cells",
"6"="cDC1",
"10"="cDC2",
"11"="Non-immune cells")
obj <- RenameIdents(obj, newIds)
# 标准化
obj <- SCTransform(obj, verbose = FALSE, variable.features.n = 10000)
DimPlot(obj) + ggplot2::coord_fixed()
② 执行geseca分析,使用geseca函数,具体可以分为如下三步:
# 1.主成分分析获得基因载荷,用于后续分析
obj <- RunPCA(obj, assay = "SCT", verbose = FALSE,
rev.pca = TRUE, reduction.name = "pca.rev",
reduction.key="PCR_", npcs = 50)
E <- obj@reductions$pca.rev@feature.loadings
# 2.从msigdbr数据库获得hallmark基因集通路
library(msigdbr)
pathwaysDF <- msigdbr("mouse", category="C2", subcategory = "CP:KEGG")
pathways <- split(pathwaysDF$gene_symbol, pathwaysDF$gs_name)
# 3.执行geseca分析
set.seed(1)
gesecaRes <- geseca(pathways, E, minSize = 5, maxSize = 500, center = FALSE, eps=1e-100)
head(gesecaRes, 10)
#> pathway pctVar pval
#> <char> <num> <num>
#> 1: KEGG_RIBOSOME 1.1844859 1.719201e-39
#> 2: KEGG_GRAFT_VERSUS_HOST_DISEASE 0.6313148 4.111787e-20
#> 3: KEGG_ALLOGRAFT_REJECTION 0.5474674 2.673392e-19
#> 4: KEGG_TYPE_I_DIABETES_MELLITUS 0.5770717 6.199822e-19
#> 5: KEGG_LEISHMANIA_INFECTION 0.4320958 1.261768e-17
#> 6: KEGG_ANTIGEN_PROCESSING_AND_PRESENTATION 0.4150349 8.298432e-17
#> 7: KEGG_AUTOIMMUNE_THYROID_DISEASE 0.6066166 1.701748e-16
#> 8: KEGG_ASTHMA 0.9968910 3.864738e-16
#> 9: KEGG_NOD_LIKE_RECEPTOR_SIGNALING_PATHWAY 0.4004829 5.916767e-16
#> 10: KEGG_LYSOSOME 0.2791275 2.030931e-15
#> padj log2err size
#> <num> <num> <int>
#> 1: 1.255017e-37 1.628072 61
#> 2: 1.500802e-18 1.151221 26
#> 3: 6.505253e-18 1.133090 24
#> 4: 1.131467e-17 1.123915 26
#> 5: 1.842182e-16 1.076868 57
#> 6: 1.009643e-15 1.057464 49
#> 7: 1.774680e-15 1.047626 21
#> 8: 3.526573e-15 1.027670 12
#> 9: 4.799155e-15 1.027670 50
#> 10: 1.482580e-14 1.007318 102
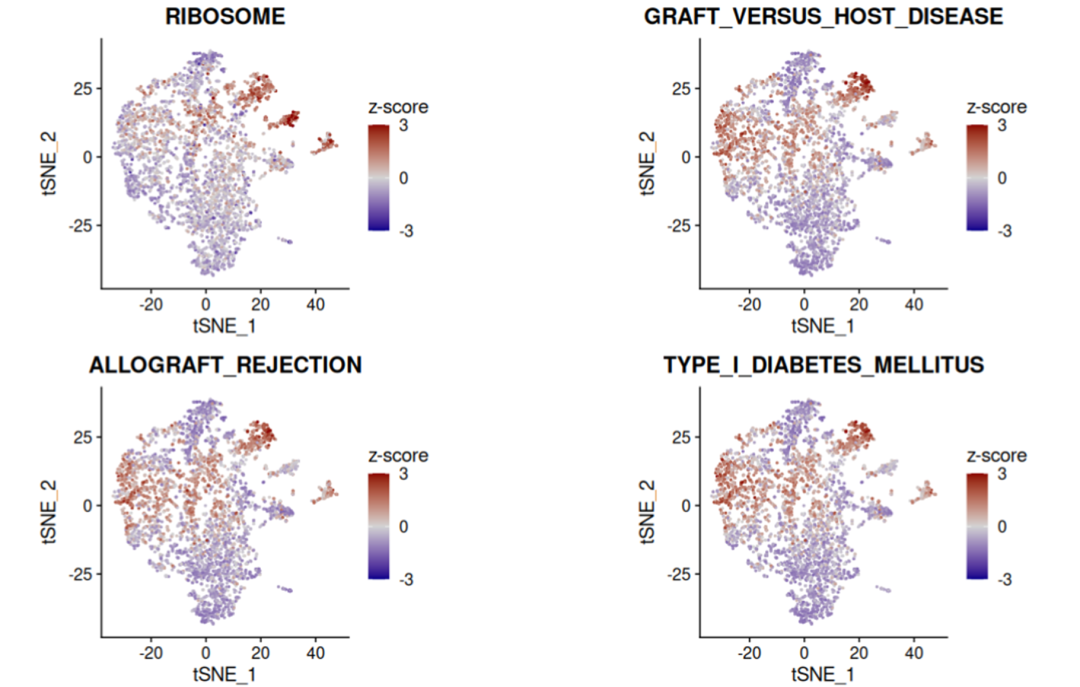
③ 绘制排名前4通路的降维图:
topPathways <- gesecaRes[, pathway] |> head(4)
titles <- sub("KEGG_", "", topPathways)
ps <- plotCoregulationProfileReduction(pathways[topPathways], obj,
title=titles,
reduction="tsne")
cowplot::plot_grid(plotlist=ps[1:4], ncol=2)
2.空转数据中的应用
① 加载空转的seurat对象,随后基于PCA提取基因载荷(和上面的流程其实是一样的):
library(Seurat)
# 读取空转数据
obj <- readRDS(url("https://alserglab.wustl.edu/files/fgsea/275_T_seurat.rds"))
# 标准化
obj <- SCTransform(obj, assay = "Spatial", verbose = FALSE, variable.features.n = 10000)
# PCA降维并收集基因载荷
obj <- RunPCA(obj, assay = "SCT", verbose = FALSE,
rev.pca = TRUE, reduction.name = "pca.rev",
reduction.key="PCR_", npcs = 50)
E <- obj@reductions$pca.rev@feature.loadings
# 使用hallmark基因集
library(msigdbr)
pathwaysDF <- msigdbr("human", category="H")
pathways <- split(pathwaysDF$gene_symbol, pathwaysDF$gs_name)
② 执行geseca分析:
set.seed(1)
gesecaRes <- geseca(pathways, E, minSize = 15, maxSize = 500, center = FALSE)
#> Warning in geseca(pathways, E, minSize = 15, maxSize = 500, center = FALSE):
#> For some pathways, in reality P-values are less than 1e-50. You can set the
#> `eps` argument to zero for better estimation.
head(gesecaRes, 10)
#> pathway pctVar pval
#> <char> <num> <num>
#> 1: HALLMARK_HYPOXIA 1.2764439 1.000000e-50
#> 2: HALLMARK_TNFA_SIGNALING_VIA_NFKB 0.5301172 2.937421e-28
#> 3: HALLMARK_EPITHELIAL_MESENCHYMAL_TRANSITION 0.4649753 3.948569e-24
#> 4: HALLMARK_INTERFERON_GAMMA_RESPONSE 0.4527210 1.198557e-23
#> 5: HALLMARK_INTERFERON_ALPHA_RESPONSE 0.4749296 1.749976e-22
#> 6: HALLMARK_OXIDATIVE_PHOSPHORYLATION 0.2667845 3.314153e-14
#> 7: HALLMARK_MTORC1_SIGNALING 0.2331203 5.824702e-12
#> 8: HALLMARK_GLYCOLYSIS 0.1878601 1.137447e-09
#> 9: HALLMARK_ALLOGRAFT_REJECTION 0.1644770 3.630902e-09
#> 10: HALLMARK_INFLAMMATORY_RESPONSE 0.1670550 1.544618e-08
#> padj log2err size
#> <num> <num> <int>
#> 1: 2.400000e-49 NA 150
#> 2: 3.524905e-27 1.3727430 151
#> 3: 3.158855e-23 1.2709130 145
#> 4: 7.191339e-23 1.2545135 150
#> 5: 8.399886e-22 1.2210538 77
#> 6: 1.325661e-13 0.9653278 181
#> 7: 1.997041e-11 0.8870750 180
#> 8: 3.412342e-09 0.7881868 144
#> 9: 9.682405e-09 0.7614608 98
#> 10: 3.707084e-08 0.7337620 112
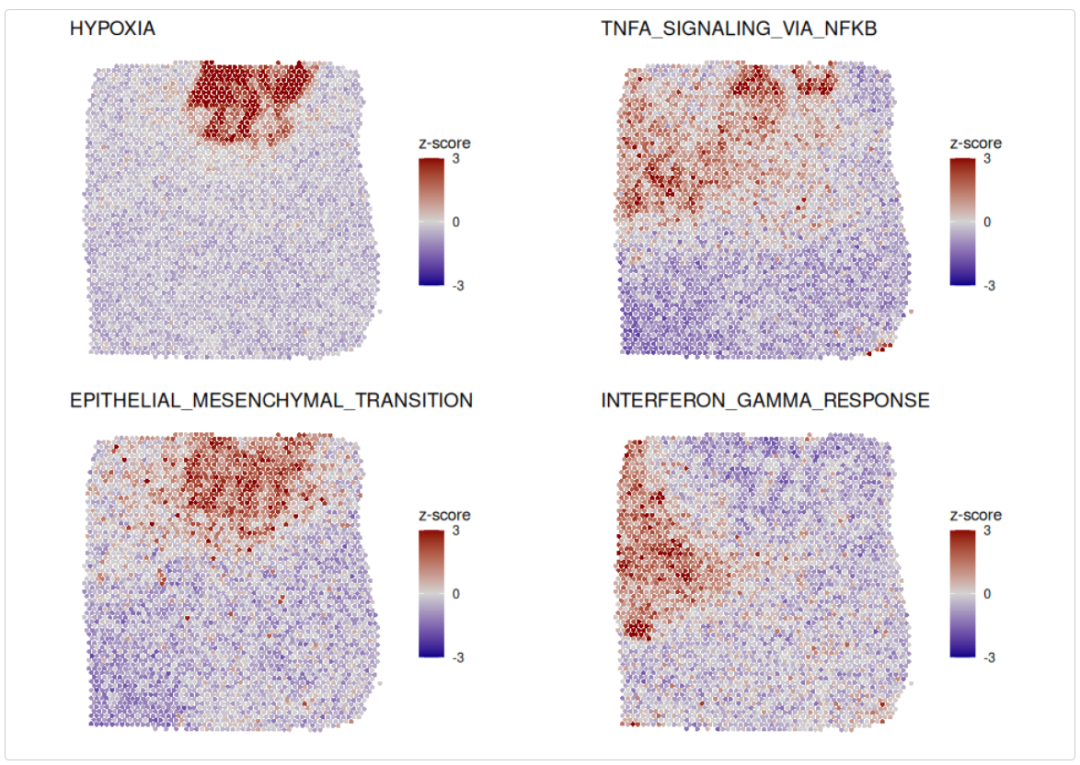
可视化前4个通路评分的空转分布:
topPathways <- gesecaRes[, pathway] |> head(4)
titles <- sub("HALLMARK_", "", topPathways)
pt.size.factor <- 1.6
# Starting from Seurat version 5.1.0, the scaling method for spatial plots was changed.
# As mentioned here: https://github.com/satijalab/seurat/issues/9049
# This code provides a workaround to adapt to the new scaling behavior.
if (packageVersion("Seurat") >= "5.1.0"){
sfactors <- ScaleFactors(obj@images$slice1)
pt.size.factor <- 1.5 * sfactors$fiducial / sfactors$hires
}
ps <- plotCoregulationProfileSpatial(pathways[topPathways], obj,
title=titles,
pt.size.factor=pt.size.factor)
cowplot::plot_grid(plotlist=ps, ncol=2)
赶紧试试呢
各位佬哥佬姐点点关注


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









