






生信碱移
Marsilea 库
复杂数据可视化神器,Marsilea。
随着数据集规模和复杂性的指数级增长,科学研究和数据分析领域对数据可视化工具提出了更高的要求。然而,传统的数据可视化工具在处理多特征、多维度数据时往往面临显著挑战,难以直观展示数据之间复杂的交互关系或揭示隐藏的模式。
为了解决这一痛点,来自澳门大学的研究者提出了 Marsilea,于2025年1月6日见刊于Genome Biology [IF:10.1]。Marsilea 是一个使用声明方式创建可组合可视化图表的Python库。它基于Matplotlib,可以像拼图一样组合不同的可视化图表。

▲ DOI: 10.1186/s13059-024-03469-3。
与之前的传统可视化工具相比,Marsilea 显著减少了代码量。例如,在对比同样的可视化任务时,Marsilea 所需的代码量只有 Matplotlib 的一半,同时提供了更高的定制性和直观性。除了代码的可视化工具以外,Marsilea还提供了在线可视化工具:
-
https://marsilea.streamlit.app/
本文主要介绍 Marsilea 的使用,想更深入了解该软件的同学可以通过以下链接学习:
-
https://marsilea.readthedocs.io/en/stable/index.html
软件安装
在安装了python的前提下,可以使用以下代码安装该库:
pip install marsilea
使用概念
① 首先导入一些必要的库:
import numpy as np
import marsilea as ma
import marsilea.plotter as mp
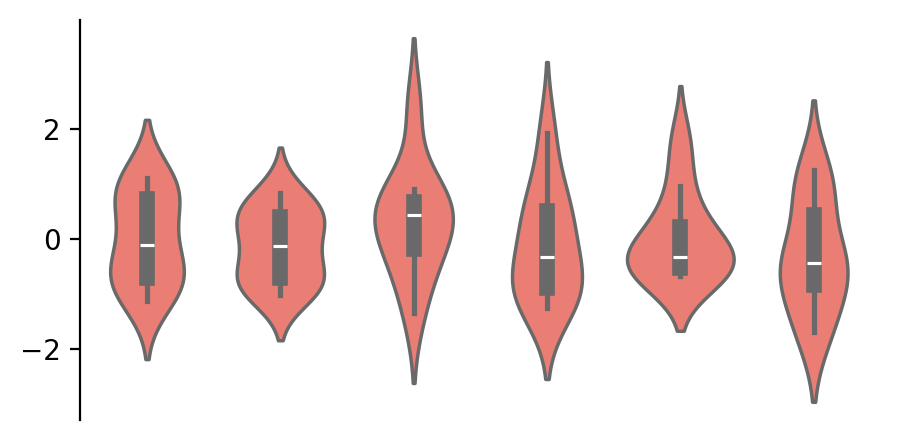
② 生成模拟数据data并创建一个图表框架:
data = np.random.randn(10, 6)
cb = ma.ClusterBoard(data, height=2, margin=0.5)
cb.add_layer(mp.Violin(data, color="#FF6D60"))
cb.render()
上面三个函数分别代表三个步骤:
-
首先,创建一个
ClusterBoard来绘制主要的可视化图形,画布的高度初始化为 2,边距为 0.5; -
其次,使用
add_layer()来在画布上添加一个 Violin 图表; -
最后,
render()被调用来显示可视化结果
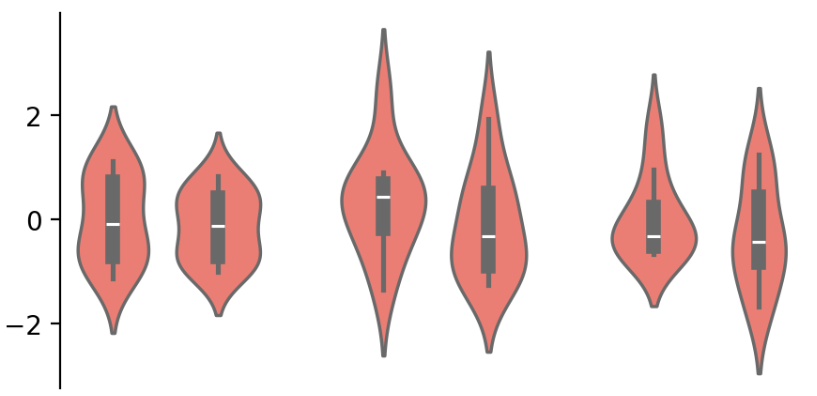
③ 创建完图表框架以后,针对示例的这个小提琴图,可以使用group_cols函数进行分组:
cb.group_cols(["c1", "c1", "c2", "c2", "c3", "c3"],
order=["c1", "c2", "c3"], spacing=.08)
cb.render()
-
group_cols()将画布分为三组; -
group 参数指定每列的组,order 参数指定将在图中呈现的组的顺序。
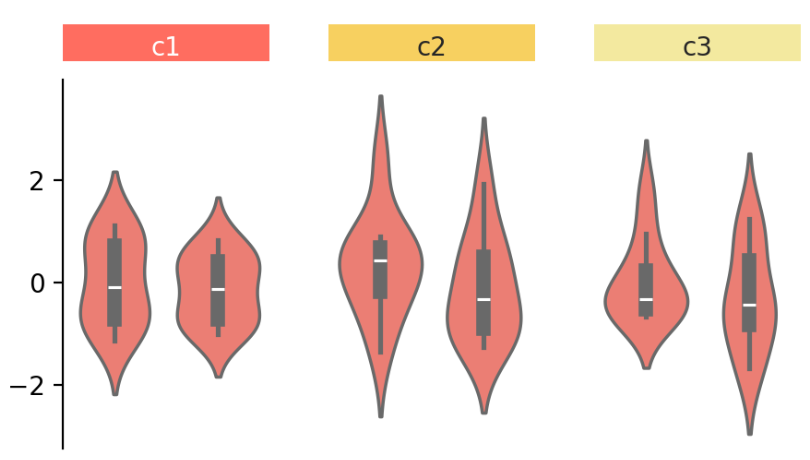
④ 随后,还可以根据分组创建注释。同样是两个命名清晰且易懂的函数:
group_labels = mp.Chunk(["c1", "c2", "c3"],
["#FF6D60", "#F7D060", "#F3E99F"])
cb.add_top(group_labels, size=.2, pad=.1)
cb.render()
-
add_top() 来在画布顶部添加一个 Chunk 图表,Chunk 图表是一个注解图表,用于标注组别;
-
此外,可以使用 size 和 pad 参数调整图表的大小和图表之间的间距
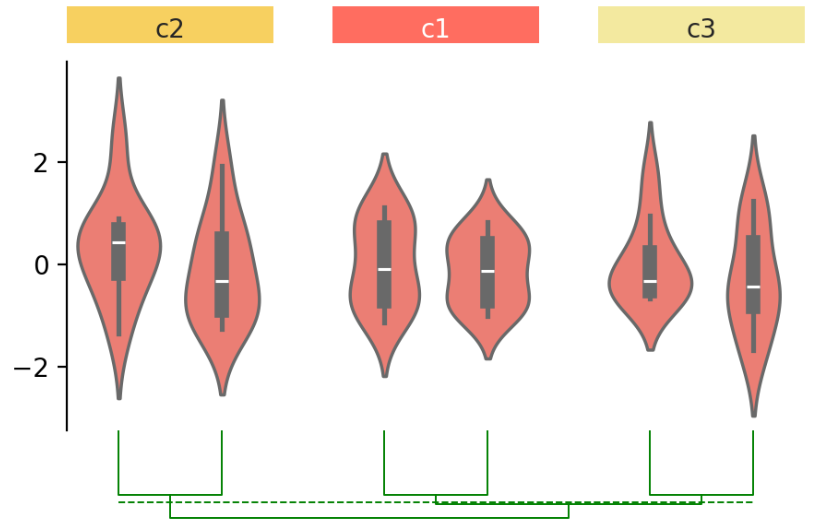
⑤ 为样本添加层次聚类:
cb.add_dendrogram("bottom", colors="g")
cb.render()
-
使用 add_dendrogram() 将 dendrogram 添加到画布底部。在 Marsilea 中,聚类可以在不同的可视化中进行,不限于热图
⑥ 最后,添加一个标题:
cb.add_bottom(ma.plotter.Bar(data, color="#577D86"), size=2, pad=.1)
cb.add_title(top="My First Marsilea Example")
cb.render()
-
使用 add_title() 在顶部添加了一个标题;
-
同时,还使用 Bar 在底部添加了一个图表。
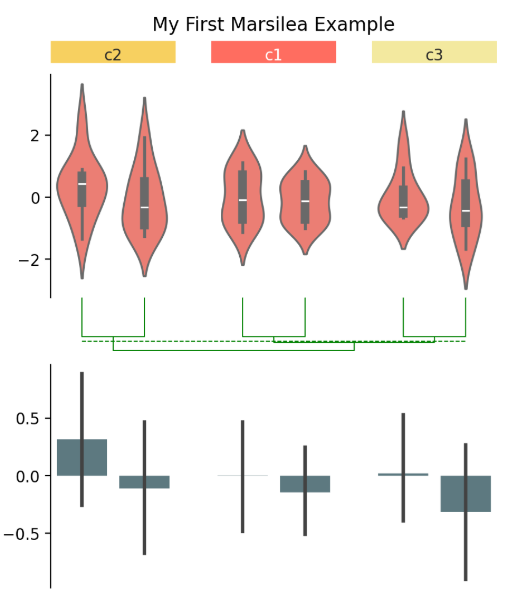
⑦ 保存生成的图片,可以像保存所有 matplotlib 图表一样保存,也可以通过.figure访问图表对象,建议以 bbox_inches="tight" 模式保存以避免裁剪:
cb.figure.savefig("my_first_marsilea_example.png", bbox_inches="tight")
真实数据集上的应用
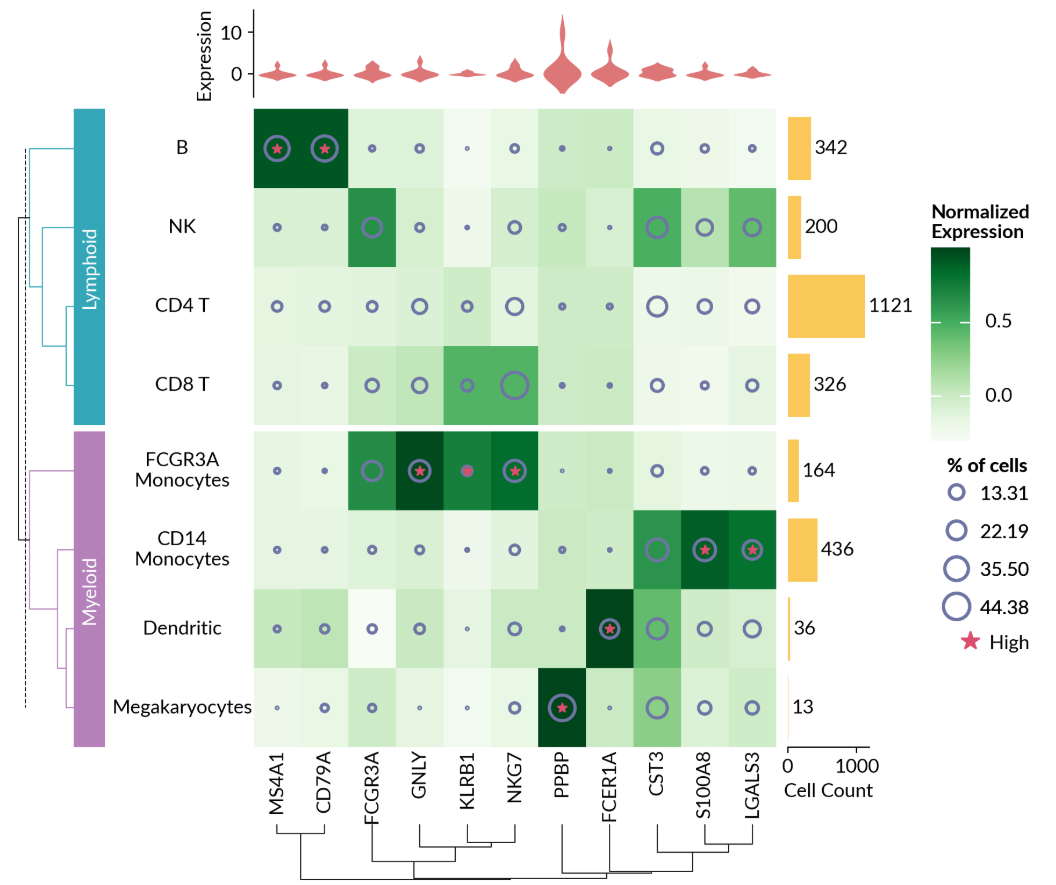
① 单细胞测序数据可视化:
import matplotlib as mpl
import matplotlib.pyplot as plt
from matplotlib.colors import Normalize
import marsilea as ma
import marsilea.plotter as mp
from sklearn.preprocessing import normalize
pbmc3k = ma.load_data("pbmc3k")
exp = pbmc3k["exp"]
pct_cells = pbmc3k["pct_cells"]
count = pbmc3k["count"]
matrix = normalize(exp.to_numpy(), axis=0)
cell_cat = [
"Lymphoid",
"Myeloid",
"Lymphoid",
"Lymphoid",
"Lymphoid",
"Myeloid",
"Myeloid",
"Myeloid",
]
cell_names = [
"CD4 T",
"CD14\nMonocytes",
"B",
"CD8 T",
"NK",
"FCGR3A\nMonocytes",
"Dendritic",
"Megakaryocytes",
]
# Make plots
cells_proportion = mp.SizedMesh(
pct_cells,
size_norm=Normalize(vmin=0, vmax=100),
color="none",
edgecolor="#6E75A4",
linewidth=2,
sizes=(1, 600),
size_legend_kws=dict(title="% of cells", show_at=[0.3, 0.5, 0.8, 1]),
)
mark_high = mp.MarkerMesh(matrix > 0.7, color="#DB4D6D", label="High")
cell_count = mp.Numbers(count["Value"], color="#fac858", label="Cell Count")
cell_exp = mp.Violin(
exp, label="Expression", linewidth=0, color="#ee6666", density_norm="count"
)
cell_types = mp.Labels(cell_names, align="center")
gene_names = mp.Labels(exp.columns)
# Group plots together
h = ma.Heatmap(
matrix, cmap="Greens", label="Normalized\nExpression", width=4.5, height=5.5
)
h.add_layer(cells_proportion)
h.add_layer(mark_high)
h.add_right(cell_count, pad=0.1, size=0.7)
h.add_top(cell_exp, pad=0.1, size=0.75, name="exp")
h.add_left(cell_types)
h.add_bottom(gene_names)
h.group_rows(cell_cat, order=["Lymphoid", "Myeloid"])
h.add_left(mp.Chunk(["Lymphoid", "Myeloid"], ["#33A6B8", "#B481BB"]), pad=0.05)
h.add_dendrogram("left", colors=["#33A6B8", "#B481BB"])
h.add_dendrogram("bottom")
h.add_legends("right", align_stacks="center", align_legends="top", pad=0.2)
h.set_margin(0.2)
h.render()
② SeqLogo可视化:
from marsilea.plotter import SeqLogo
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
matrix = pd.DataFrame(data=np.random.randint(1, 10, (4, 10)), index=list("ACGT"))
_, ax = plt.subplots()
colors = {"A": "r", "C": "b", "G": "g", "T": "black"}
SeqLogo(matrix, color_encode=colors).render(ax)

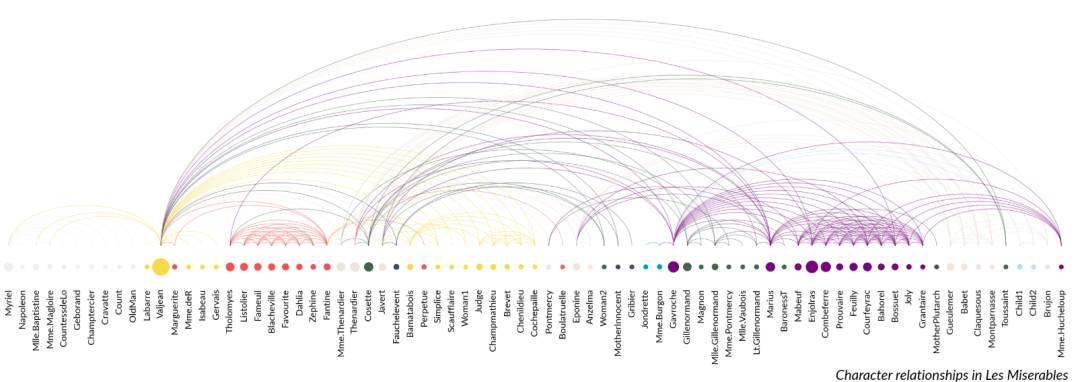
③ 网络弧图:
import marsilea as ma
import marsilea.plotter as mp
data = ma.load_data("les_miserables")
nodes = data["nodes"]
links = data["links"]
sizes = nodes["value"].to_numpy().reshape(1, -1)
colors = nodes["group"].to_numpy().reshape(1, -1)
palette = {
0: "#3C486B",
1: "#F0F0F0",
2: "#F9D949",
3: "#F45050",
4: "#F2E3DB",
5: "#41644A",
6: "#E86A33",
7: "#009FBD",
8: "#77037B",
9: "#4F4557",
10: "#B0DAFF",
}
link_colors = [palette[nodes.iloc[i].group] for i in links["source"]]
height = 0.5
width = height * len(nodes) / 3
sh = ma.SizedHeatmap(
sizes,
colors,
palette=palette,
sizes=(10, 200),
frameon=False,
height=height,
width=width,
)
sh.add_bottom(mp.Labels(nodes["name"], fontsize=8))
arc = mp.Arc(nodes.index, links.to_numpy(), colors=link_colors, lw=0.5, alpha=0.5)
sh.add_top(arc, size=3)
sh.add_title(
bottom="Character relationships in Les Miserables",
align="right",
fontstyle="italic",
)
sh.render()
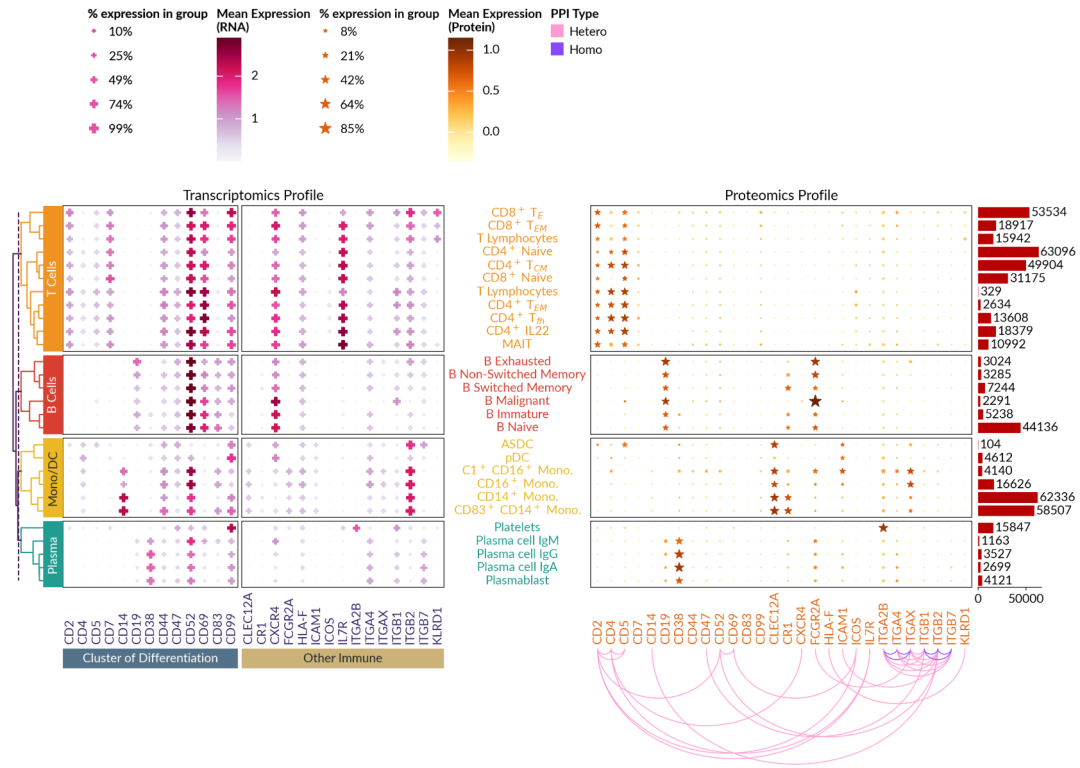
④ 单细胞多组学数据可视化:
import matplotlib as mpl
from matplotlib.ticker import FuncFormatter
import marsilea as ma
dataset = ma.load_data("sc_multiomics")
fmt = FuncFormatter(lambda x, _: f"{x:.0%}")
lineage = ["B Cells", "T Cells", "Mono/DC", "Plasma"]
lineage_colors = ["#D83F31", "#EE9322", "#E9B824", "#219C90"]
m = dict(zip(lineage, lineage_colors))
cluster_data = dataset["gene_exp_matrix"]
interaction = dataset["interaction"]
lineage_cells = dataset["lineage_cells"]
marker_names = dataset["marker_names"]
cells_count = dataset["cells_count"]
display_cells = dataset["display_cells"]
with mpl.rc_context({"font.size": 14}):
gene_profile = ma.SizedHeatmap(
dataset["gene_pct_matrix"],
color=dataset["gene_exp_matrix"],
height=6,
width=6,
cluster_data=cluster_data,
marker="P",
cmap="PuRd",
sizes=(1, 100),
color_legend_kws={"title": "Mean Expression\n(RNA)"},
size_legend_kws={
"colors": "#e252a4",
"fmt": fmt,
"title": "% expression in group",
},
)
gene_profile.group_rows(lineage_cells, order=lineage)
gene_profile.add_left(ma.plotter.Chunk(lineage, lineage_colors, padding=10))
gene_profile.add_dendrogram(
"left",
method="ward",
colors=lineage_colors,
meta_color="#451952",
linewidth=1.5,
)
gene_profile.add_bottom(
ma.plotter.Labels(marker_names, color="#392467", align="bottom", padding=10)
)
gene_profile.cut_cols([13])
gene_profile.add_bottom(
ma.plotter.Chunk(
["Cluster of Differentiation", "Other Immune"],
["#537188", "#CBB279"],
padding=10,
)
)
gene_profile.add_right(
ma.plotter.Labels(
display_cells,
text_props={"color": [m[c] for c in lineage_cells]},
align="center",
padding=10,
)
)
gene_profile.add_title("Transcriptomics Profile", fontsize=16)
protein_profile = ma.SizedHeatmap(
dataset["protein_pct_matrix"],
color=dataset["protein_exp_matrix"],
cluster_data=cluster_data,
marker="*",
cmap="YlOrBr",
height=6,
width=6,
color_legend_kws={"title": "Mean Expression\n(Protein)"},
size_legend_kws={
"colors": "#de600c",
"fmt": fmt,
"title": "% expression in group",
},
)
protein_profile.group_rows(lineage_cells, order=lineage)
protein_profile.add_bottom(
ma.plotter.Labels(marker_names, color="#E36414", align="bottom", padding=10)
)
protein_profile.add_dendrogram("left", method="ward", show=False)
score = interaction["STRING Score"]
protein_profile.add_bottom(
ma.plotter.Arc(
marker_names,
interaction[["N1", "N2"]].values,
# weights=score,
colors=interaction["Type"].map({"Homo": "#864AF9", "Hetero": "#FF9BD2"}),
labels=interaction["Type"],
width=1,
legend_kws={"title": "PPI Type"},
),
size=2,
)
protein_profile.add_right(ma.plotter.Numbers(cells_count, color="#B80000"), pad=0.1)
protein_profile.add_title("Proteomics Profile", fontsize=16)
comb = gene_profile + protein_profile
comb.add_legends("top", stack_size=1, stack_by="column", align_stacks="top")
comb.render()
虽然complexheatmap也有py版
但是感觉这个工具也蛮好用的


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









