







 文章介绍了生物信息学如何通过比较氨基酸序列来洞察蛋白质的结构和功能,包括序列比对、进化树构建、序列变化类型,以及类似序列在预测结构和功能上的重要性。此外,讨论了氨基酸交换频率和BLOSUM矩阵在蛋白质序列比对中的作用。
文章介绍了生物信息学如何通过比较氨基酸序列来洞察蛋白质的结构和功能,包括序列比对、进化树构建、序列变化类型,以及类似序列在预测结构和功能上的重要性。此外,讨论了氨基酸交换频率和BLOSUM矩阵在蛋白质序列比对中的作用。
《Protein Actions Principles and Modeling》-《蛋白质作用原理和建模》
本人能力有限,如果错误欢迎批评指正。
第八章 Bioinformatics: Insights from Protein Sequences
(生物信息学:来自蛋白质序列的见解)
-比较氨基酸序列可以深入了解蛋白质的结构和功能
生物信息学方法可以用于比较和分析蛋白质或核酸的序列,从而了解它们的功能。给定一个氨基酸序列,人们通常希望在数据库中找到一个类似的序列。这些搜索被称为字符串搜索。有时,您希望对齐和比较两个或多个序列,并测量它们之间的距离。通过测量序列之间的距离,我们可以构建进化树,或者推断生物学功能或结合位点,或者了解哪些药物可能与这些蛋白质结合。通过观察在进化过程中保守的序列位置可以推断出蛋白质的稳定性或功能。而且,如果你正在设计一种药物来禁用致病性生物体,你可以将病原体蛋白和人类蛋白质进行比较,以避免错误地给人类蛋白质用药,导致药物的副作用。
第一个对蛋白质完整氨基酸序列的实验测定是胰岛素序列,由F Sanger于1951年发表。为此,他在1958年获得了诺贝尔化学奖。在20世纪60年代,M Dayhoff在一个已发表的数据库中组装了第一个蛋白质序列数据集,在20世纪60年代,M Dayhoff在一个已发表的数据库中组装了第一个蛋白质序列数据集,并将其称为 Atlas of Protein Sequence and Structure。她开发了第一个算法,根据序列的相似性将其聚类为家族和超家族。我们对蛋白质结构的了解从1960年的2人增长到2016年的12万(其中超过10万是由x射线晶体学确定的,1万是由核磁共振确定的,几千种是由低温电子显微镜确定的)。序列数据库增长得甚至更快(图8.1)。例如GenBank,它是一个公开的DNA序列数据库。而UniProtKB(通用蛋白质知识库),它是一个蛋白质序列数据库。图8.1还显示了基于CATH分类数据库的蛋白质超家族总数的增长,当有足够的证据表明蛋白质结构域与共同祖先分化时,它们被划分为超家族。
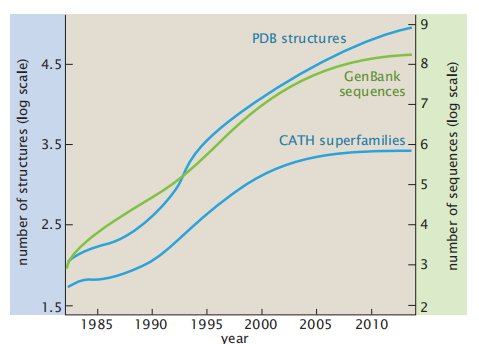
图8.1 已知序列和结构的数量增长。来自GenBank的序列(右纵坐标)。来自PDB的结构(左纵坐标)。然而,新的蛋白质超家族的数量正在达到一个平台期。CATH数据库版本v4.1包含2737个超家族。
序列通过进化突变而改变
进化导致蛋白质序列中的氨基酸变化。有三种类型的序列变化起源于DNA的变化:突变、插入和缺失(mutations, insertions, and deletions)(图8.2)。在突变中,一个氨基酸被交换为另一个;在缺失中,一个氨基酸被删除;在插入中,一个额外的氨基酸被添加,因为一个父序列进化成一个子序列。Indel是一个术语,指发生插入或删除的情况。
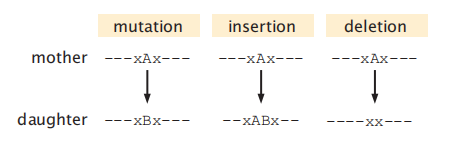
图8.2 不同类型的突变变化:替换、插入和删除。
具有相似序列的蛋白质通常具有相似的结构和功能
如果两个蛋白质具有相似的氨基酸序列,它们可能也具有相似的天然结构和相似的生物学功能和作用机制。事实上,结构的发散比序列要慢,许多序列可能具有相同的结构,如第7章中的图7.10所示。因此,我们可以利用氨基酸序列的相似性和差异性来推断生物学中的结论。这是生物信息学和比较建模的主题(bioinformatics and comparative modeling),在这里,我们可以通过比较生物分子的序列来推断它们的各种特性。
当两个序列彼此仅稍微相似时,它们可以被称为位于序列相似性的过渡地带(twilight zone)。非常相似的序列都在可靠区域(reliable zone)内。而且,如此不同以至于序列-功能关系不可预测的序列则在午夜区域(midnight zone)。过渡地带是指序列有20-35%相同的地方(图8.3)。两个序列同源性大于35%的蛋白质很可能具有相似的结构和执行相似的功能。
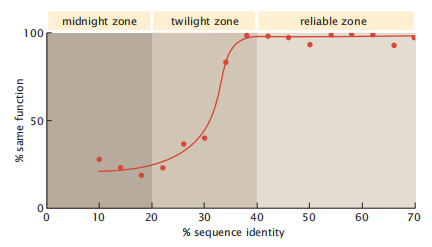
图8.3 两个具有相似序列的蛋白质通常具有相似的功能。高序列同一性(可靠区)意味着两个蛋白质具有相同的功能。但有时蛋白质具有相同的功能,即使它们没有高序列的一致性(午夜区)。在这两者之间(过渡带),序列对功能的可预测性较低。
-你如何确定序列之间的相关性?

图8.4 一个亲本序列可以进化为不同的子序列。左边的母女对序列有一个突变。右边的母女对有一个缺失(在A的第2位),一个突变(在第6位,从M到R)和一个插入(T,在第7位)。然而,要确定哪一对母女关系更密切,你需要做的不仅仅是数它们;您还需要对每个修改的重要性进行评分。
图8.4 显示了两对不同的父子序列。每个女儿和父母有多关系?哪对母女的“关系更亲密”?左边的那一对有一个突变。右边的一对有一个缺失,一个突变和一个插入。那么,左边的这对关系更密切吗?不一定。为了度量亲缘关系,您需要一个度量标准,也就是说,这是一种对任何给定的突变、插入或删除的“价值”进行评分的方法。两个序列之间的相关性程度取决于您如何选择对任何特定类型的变化的重要性进行排序。由此可见,对评分系统的选择存在一定的任意性。以下是确定两个序列之间的相似性的步骤。
首先,要在序列之间进行比较,您必须将它们彼此对齐(参见第186页)。对齐序列意味着将两个序列叠加,以找到它们的子串之间的共性、就像在一个被损坏的信息传输中找到单词和句子一样。图8.5 给出了一个序列对齐的示例。
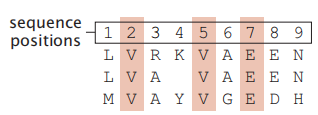
图8.5 对齐后的序列在某些位置上有相同的残基。在这三个序列的比对中,在序列位置2、5和7上有一个相同的残基。对齐有时需要跳过一个位点(一个删除),在这种情况下是在中间序列的第4位点。
其次,对于两个对齐的序列,我们需要计算一个分数,用于表明一个序列与另一个序列的相似程度。我们通常使用简约原则(parsimony principle,),即从祖先到后代的变化应该是与序列差异相一致的最小数字来计算这个分数。如果父系中看到残基X,同时在一个祖先中看到相同位置的残基Y,那么我们就假设从X到Y只发生了一个突变。在现实中,可能有很多变化,X→Z→W→Y,但如果对这些变化没有直接的了解,我们应该假设最小的变化。此外,您还可以为父级或祖先级中的间隙分配一些惩罚分数,即为indels分配一些间隙。根据不同的间隙长度和其他属性,不同的算法以不同的方式对间隙进行评分。一个通常用于为k个残基的间隙分配一个惩罚g (k)的表达式是:

其中a是表示间隙起始成本(gap-initiation cost)的常数,b是间隙扩展成本(gap-extension cost),对间隙中的每个氨基酸平均惩罚。公式8.1表示,所有间隙都有内在的起始成本a,并且总惩罚与间隙的大小成线性比例变得更高。通常设置b < a,表示打开一个间隙的惩罚大于延长一个额外氨基酸的惩罚。通常使用像= 11和b = 1这样的值来体现一个通用的观点,即如果两个序列的长度较短,那么两个序列在进化上关系更紧密。
Hamming距离(见第7章的方框7.1)很简单,只对每个位置进行二进制确定:要么两个序列完全匹配,要么不匹配。在实践中,评分系统通常比这更精细。例如,可以根据物理尺度对一个氨基酸交换为另一个氨基酸进行评分,例如它们的疏水性、电荷或大小(参见第1章中的图1.5)。例如,亮氨酸通常可以交换缬氨酸,因为它们既疏水又大小相同。相比之下,半胱氨酸和谷氨酸很少交换,因为它们的物理性质彼此不同。
有些氨基酸比其他氨基酸交换得更频繁
比较多个相关的蛋白质时,我们会看到不同的不同位点的氨基酸分布。我们想确定氨基酸的交换频率。要做到这一点,可以通过首先配对两个已知是同源物且具有很高序列相似性的蛋白质来建立一个数据库。然后,我们对齐它们的序列-对多个蛋白对做这样的操作。然后,计算在序列A中i位置a型氨基酸和在同源序列A'中同一i位置找到b型氨基酸的次数,获得频率pab(i)。接下来,为了将其转换为一个有用的交换度量,需要进行两个数学计算。首先,你要对随机发现的东西进行修正。设qa为a型氨基酸出现的自然频率(a型氨基酸的数量除以在大量的蛋白质中所有类型氨基酸的数量)。然后,将序列位置i的对数比值分数sab (i)定义为

当pab (i)/(qaqb)= 1时,这意味着a型和b型氨基酸在i位置的交换速率与偶然预期的交换速率相同,然后sab (i) = 0。当pab (i)/(qaqb)> 1时,这意味着氨基酸类型a和b交换的频率高于随机(所以sab (i) > 0),而当pab (i)/(qaqb)< 1时,这意味着它们交换的频率低于随机(所以sab (i) < 0)。在方程8.2中取对数的原因是,它使数学更方便地评分完整的氨基酸序列,而不仅仅是单个位置。比较两个完整序列A和A'的总整体得分是所有位点i上的sab(i)的总和。这种方法,以及它的变体(使用大量的蛋白质集和取平均值),是氨基酸替代矩阵的基础(Box 8.1),它可以分配分数来找到最佳的序列比对。在蛋白质序列比对中常用的一种替代矩阵是BLOSUM。
====================================================
Box 8.1 替换矩阵的例子:BLOSUM和PSSM
BLOSUM(BLOck子替代矩阵)是为了改进进化上遥远的序列的比较。BLOSUM62如图8.6所示(62表示分数来自于比较62%或以下的序列)。
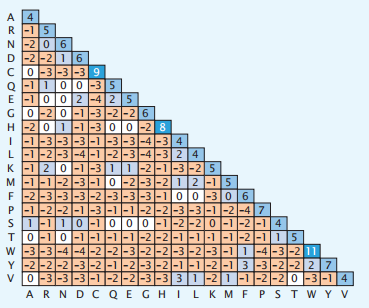
图8.6 BLOSUM62相似度矩阵。这个矩阵中的每个矩阵元素都是用一种特定氨基酸替代另一种氨基酸的对数比值比(见等式8.2)。这个矩阵是对称的,所以只显示了一半。非对角线元素描述了交换两种不同类型的氨基酸的倾向。正数意味着这些互换发生的频率比偶然的更高,而负数则表明它们比偶然的更罕见。对角线项表示每种氨基酸的保守程度。例如,色氨酸、半胱氨酸和组氨酸是最保守的。
PSSM(位置特异性替代矩阵)。之前,我们描述了替代频率只取决于氨基酸的a和b类型,而不依赖于它们在蛋白质序列中的特定位置的方法。然而,一般来说,同源蛋白之间的氨基酸替换频率可能因序列中的一个位点而不同(因此在蛋白质的天然结构中也是不同)。PSSM是一个单独解释每个残差位置i的矩阵。
====================================================
-------------------------------------------
欢迎点赞收藏转发!
下次见!


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









