










前文链接:
图形化开放式生信分析系统开发 - 1 需求分析及技术实现
图形化开放式生信分析系统开发 - 2 样本信息处理
图形化开放式生信分析系统开发 - 3 生信分析流程的进化
图形化开放式生信分析系统开发 - 4 生信分析流程的图形化
图形化开放式生信分析系统开发 - 5 生信分析流程服务器端运行
图形化开放式生信分析系统开发 - 6 生信分析流程批量运行与过程控制
图形化开放式生信分析系统开发 - 7 分析报告的模板定制与自动生成
图形化开放式生信分析系统开发 - 8 软件稳定性测试
前文讲述了从样本信息录入,分析流程的搭建/设计和运行,最终模板报告的定制与自动生成。为了实现完整的自动化,本文讲述如何与Illumina测序仪衔接,实现下机数据自动拆分(测试过的机型MiSeq,NextSeq500)。
软件获取:官网sliverworkspace.com免费下载个人版,最新版本 2.0.277363
一、bcl2fastq安装与运行
说到Illumina的下机数据拆分,就一定会用到Illumina官方的软件bcl2fastq2,目前最新版本2.20
软件可以从Illumina官网下载,默认提供的是Linux rpm格式或者是源码格式。
安装
- CentOS/Redhat:
rpm -install -y bcl2fastq2_0v2.20.0.422-2_amd64.rpm
- Ubuntu 可以将rpm转换为deb包格式然后安装:
sudo apt-get install alien
sudo alien bcl2fastq2_0v2.20.0.422-2_amd64.rpm
dpkg -i bcl2fastq2_0v2.20.0.422-2_amd64.deb
-源码安装:
## For example, you can set the environment variables as:
export TMP=/tmp
export SOURCE=${TMP}/bcl2fastq
export BUILD=${TMP}/bcl2fastq2-v2.19.x-build
export INSTALL_DIR=/usr/local/bcl2fastq2-v2.19.x
##Build and Installthe Software
${TMP}/bcl2fastq:
cd ${TMP}
tar -xvzf bcl2fastq2-v2.19.x.tar.gz
mkdir ${BUILD}
cd ${BUILD}
chmod ugo+x ${SOURCE}/src/configure
chmod ugo+x ${SOURCE}/src/cmake/bootstrap/installCmake.sh
${SOURCE}/src/configure --prefix=${INSTALL_DIR}
cd ${BUILD}
make
make install
Convert and Demultiplex BCL Files
用<runfolder>表示下机目录,默认用于标记样本的SampleSheet.csv文件位置<runfolder>/SampleSheet.csv
运行:
nohub nohup /usr/local/bin/bcl2fastq \
--runfolder-dir <RunFolder>
--output-dir <BaseCalls>
--sample-sheet <runfolder-dir>/SampleSheet.csv
或者指定SampleSheet.csv文件位置:
nohub nohup /usr/local/bin/bcl2fastq \
--runfolder-dir <RunFolder>
--output-dir <BaseCalls>
实际运行命令举例:
bcl2fastq -r 8 -p 8 -w 8 \
--no-lane-splitting \
--barcode-mismatches 1 \
--runfolder-dir /opt/ref/data/181105_NB551525_0003_AH7W7TAFXY \
--sample-sheet /opt/ref/data/181105_NB551525_0003_AH7W7TAFXY/SampleSheet.csv \
--output-dir /opt/ref/data
bcl2fastq命令参数很多,但是最常用的参数就以下几个:
- -r, --loading-threads 加载线程数
- -p, --processing-threads 拆分数据线程数
- -w, --writing-threads 数据写入线程数
- –no-lane-splitting 不按照lane拆分数据
- –barcode-mismatches 允许的barcode错配数
- –runfolder-dir / -R 下机数据目录
- –sample-sheet samplesheet文件所在位置
- –outputdir / -o 输出目录
二、如何识别下机数据目录
Illumina测序仪下机数据文件夹命名: YYMMDD_machinename_XXXX_FCXXXX
- YYMMDD 为上机日期
- machinename为该测序仪唯一命名编号
- XXXX为expirement ID
- FCXXXX为flowcell 编号
如果要使用程序扫描匹配样本系统,就需要提供至少前两个字段作为识别匹配。后两个字段,获取较难。
如何判断测序结束?一般使用该目录下RTAComplete.txt是否存在来判断测序是否完成。
三、SampleSheet.csv文件格式
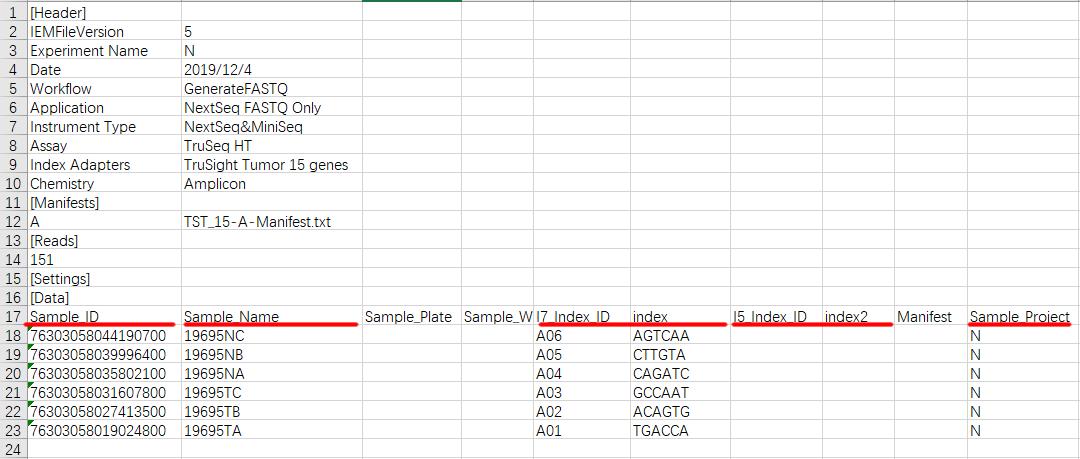
SampleSheet文件最常用的有版本4 / 5,字段有些差异,但是真正在数据拆分时候起作用的,上图红色部分,其余并不重要。这里为了拆分项目通用性使用了字段 SampleProject,实际使用种大概率会出现多个项目上一张芯片。
以上SampleSheet.csv文件放在下机数据目录里,数据分拆后得到的数据是这样的:
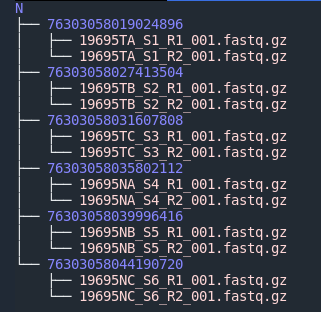
目录结构为:
[Sample Project]/[Sample ID]/[Sample Name]S*_R1_*.fastq.gz
记住此目录结构,为了拆分数据得到的数据与后续生信pipeline连接做全自动分析。
四、与系统交互从样本信息中生成SampleSheet.csv
如果要用程序生成SampleSheet文件,这里就会用到图形化开放式生信分析系统开发 - 2 样本信息处理文章里样本信息的字段信息。
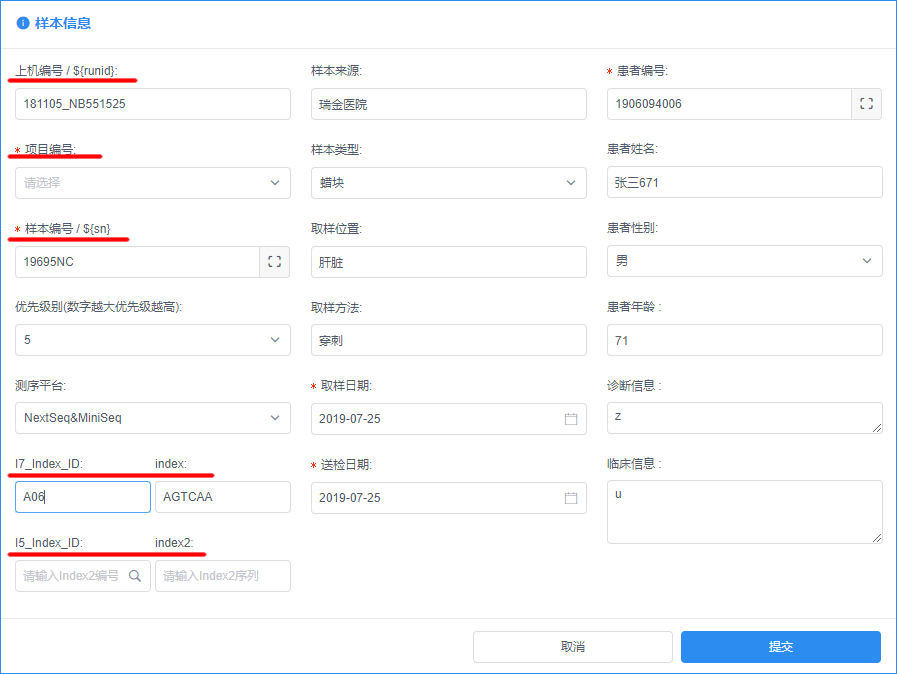
- 上机编号即对应于Illumina测序仪下机数据目录,前两个字段
五、与分析流程对接,实现拆分数据与数据分析联动
需要完成的工作:
-
请求系统根据样本信息生成SampleSheet,并下载到本地下机数据目录。
-
运行bcl2fastq分拆数据。
-
分拆成功后,更新系统中相关的Sample状态,标识该样本数据已经分拆过,避免重复运行。
-
在pipeline起始输入端,匹配分拆后的数据输入目录。必要时候使用正则表达式匹配。
如:
${pn}/${id}/^${sn}_S[a-zA-Z0-9_.\-]+_R1_[0-9]+.fastq.gz$
${pn}/${id}/^${sn}_S[a-zA-Z0-9_.\-]+_R2_[0-9]+.fastq.gz$
整体的运行逻辑流程图如下:
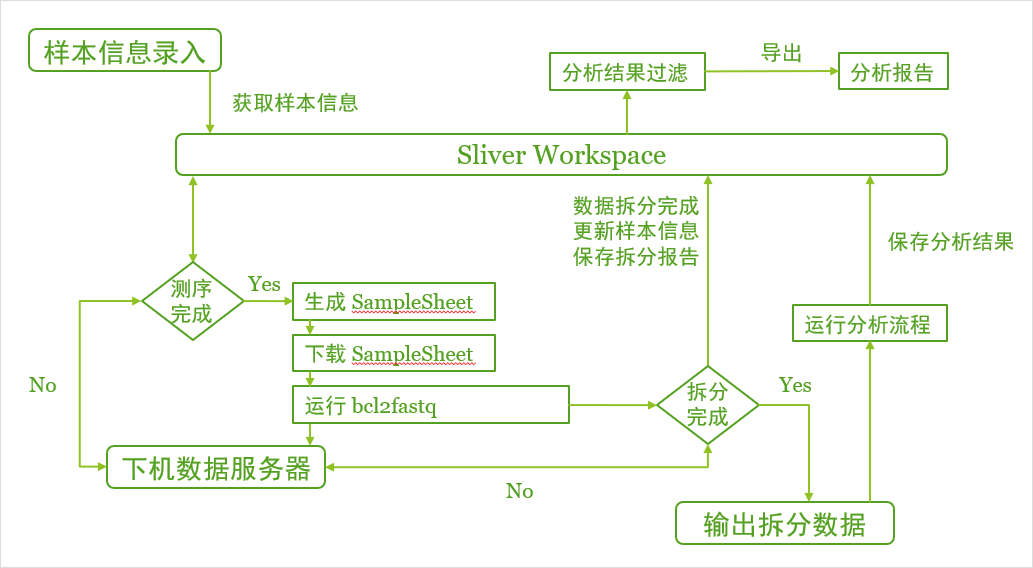
六、拆分运行日志与报告保存
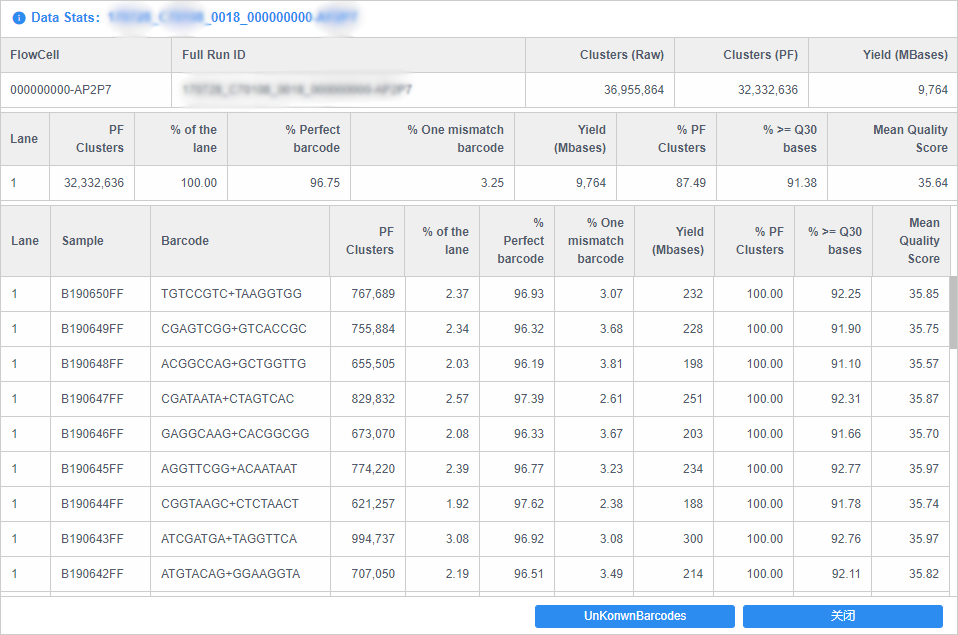
单纯的体力劳动。上两张图
下载产品PPT 或加QQ群:853718264参与讨论

















 223
223

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









