






 本文介绍了使用VASP进行表面吸附计算的过程,以W(100)表面吸附CO分子为例,详细阐述了从结构优化、吸附能计算到单原子和多原子催化剂稳定性的计算方法,提供了关键的INCAR设置和能量计算公式。
本文介绍了使用VASP进行表面吸附计算的过程,以W(100)表面吸附CO分子为例,详细阐述了从结构优化、吸附能计算到单原子和多原子催化剂稳定性的计算方法,提供了关键的INCAR设置和能量计算公式。
本文主要为了记录在学习Vasp计算过程中如何设置INCAR中的一些参数,并不着重讲解其含义,详见可自行到vasp官网查阅。
其次关于建模部分也不做细致讨论,一般结构可从Materials Studio里自带的晶体数据库导出,或从三大数据库网站上下载:
1、Materaials Project网址:https://materialsproject.org/(强烈推荐,邮箱注册即可使用)
2、CCDC网址:https://www.ccdc.cam.ac.uk/
3、ISCD:http://www2.fiz-karlsruhe.de/icsd_home.html
一、表面吸附计算
这里以W(100)表面吸附CO分子为例,暂不做收敛性测试,主要为了记录一些参数的设定,计算流程如下:
1.对W晶胞进行结构优化
INCAR设置:
#### initial I/O ####SYSTEM = WISTART = 0 ICHARG = 2 LWAVE = .FALSE. #优化晶胞时,不开波函数,节省计算时间LCHARG = .FALSE. #优化晶胞时,不开电荷密度,节省计算时间#### Ele Relax 电子步#### ENCUT = 225 #截断动能根据相关文献或赝势确定,也可自己做收敛性测试ISMEAR = 1 #金属体系SIGMA = 0.2 #POTCAR值确定EDIFF = 0.1E-4 NELM = 60 LREAL = .F.PREC = Normal#### Geo opt 离子步####EDIFFG = -0.05IBRION = 2 POTIM = 0.2 NSW = 100 ISIF = 3 #需要优化W的离子位置、晶胞常数和体积POSCAR设置(固定原子优化):
W bulk1.0 3.16520000 0.00000000 0.00000000 0.00000000 3.16520000 0.00000000 0.00000000 0.00000000 3.16520000 W2Selective dynamics #固定原子位置优化时添加Direct 0.000000000 0.000000000 0.000000000 F F F #F代表固定该原子位置 0.500000000 0.500000000 0.500000000 F F F #T代表放开优化该原子2.W晶胞切W(100)面并优化
INCAR设置:
#### initial I/O ####SYSTEM = WISTART = 0 ICHARG = 2 LWAVE = .FALSE. LCHARG = .FALSE. #### Ele Relax #### ENCUT = 225 ISMEAR = 1 SIGMA = 0.2 EDIFF = 0.1E-4 NELM = 60 LREAL = .F.PREC = NormalALGO = Fast#### Geo opt ####EDIFFG = -0.05IBRION = 2 POTIM = 0.2 NSW = 100 ISIF = 2 #优化表面3.优化CO分子
- 将CO分子放置在bulk中,晶格常数:a=b=c=10;α=β=γ=90
INCAR设置:
#### initial I/O ####SYSTEM = COISTART = 0 ICHARG = 2 LWAVE = .F. LCHARG = .F. #### Ele Relax #### ENCUT = 400 ISMEAR = 0 SIGMA = 0.2 EDIFF = 0.1E-4 NELM = 60 LREAL = .F.ALGO = Normal#### Geo opt ####EDIFFG = -0.02IBRION = 2 POTIM = 0.2 NSW = 100 ISIF = 24.W(100)表面上吸附CO分子并优化
- CO分子放置在W(100)表面上的bridge位置上,C-W键长1.90A,C-O键长1.144A。
- INCAR设置同上,只需修改POTCAR和K点即可。
5.CO吸附能计算
- 从OUTCAR文件中可以得到W(100)-CO的Total energy为:
- CO的能量为:
- W(100)表面的能量为:
- 计算吸附能公式如下:
得到CO分子的吸附能
6.如何计算单原子催化剂的结合能
- 结合能公式如下:
其中,是负载了单原子的整体催化剂能量: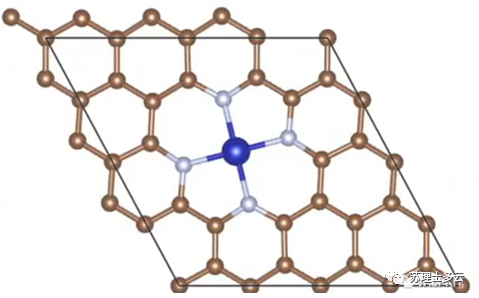 是没有负载单原子时,载体的能量:
是没有负载单原子时,载体的能量: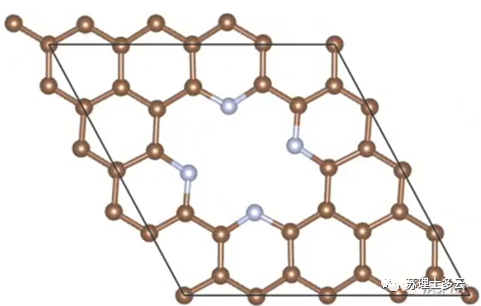 是每个单原子的能量,是用无限大的块体金属中平均每个金属的能量:
是每个单原子的能量,是用无限大的块体金属中平均每个金属的能量: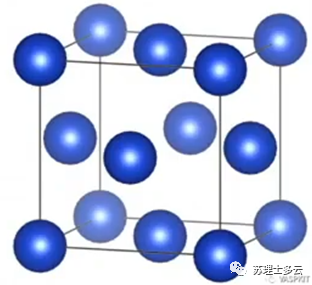
: 为什么单原子催化剂的结合能往往算出来是正值?(正值代表吸热)
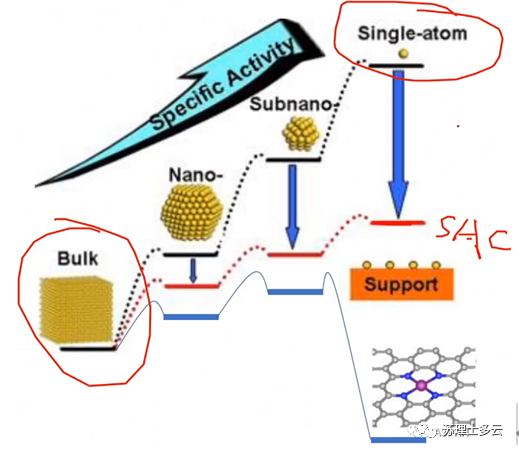 答:因为我们用上述方法算出的结合能是和Bulk能量比较,所以一般算出来是正值,除了负载在石墨烯上的单原子具有较强的稳定性其结合能为负值;如果是和孤立的单原子比较的话其结合能就是负值了。
答:因为我们用上述方法算出的结合能是和Bulk能量比较,所以一般算出来是正值,除了负载在石墨烯上的单原子具有较强的稳定性其结合能为负值;如果是和孤立的单原子比较的话其结合能就是负值了。:为啥不用下面的模型计算单原子的能量,也就是真空原子的能量。有以下两点原因(1、化学意义;2、算不准)
 答:
答:
(1)化学意义。为了解释这个问题,我们还要从化学本质上去考虑。单原子催化剂最终会失活发生聚集,聚集产物就是大的金属颗粒,那么为了探讨单原子催化剂的稳定性,肯定要和大块金属去比,而不是和真空中的单原子去比。(2)算不准,对于Cu原子还好,但是对于Fe,Co,Ni这些孤立原子,不同的初始磁矩设置会得到非常不一样的结果(差1eV以上)。这样怎么让别人去重复这些结果呢?怎么保证我们计算是有意义的?如果是块体金属,Fe,Co,Ni也有实验的磁矩作为参考,最后大家算出来的结果都是可靠、可重复的。DFT是更擅长横向比较的。
7.双原子催化剂的稳定性计算
公式同单原子基本一样:
8.n个原子的团簇的稳定性计算
这时候有两种方案都可以了,因为随着原子数增多,这个时候DFTR计算对于团簇会给出一个比较可靠的能量值了(可重复)
以上内容均为学习中的一些总结,如有错误,还请批评指教。

















 4508
4508

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









