




注:本文为“基因(Gene)”相关合辑。
略作重排,如有内容异常,请看原文。
基因(Gene)| 科学命名 / DNA 中的禁令
一、“基因”(Gene)一词的科学溯源
1.孟德尔的遗传因子探索:19 世纪中期,孟德尔通过豌豆杂交实验发现遗传规律,首次用 “天性”(Anlage)和 “因子”(Elemente)描述控制性状的遗传物质,前者指细胞内整体遗传物质,后者特指单个基因。
2.科伦斯与德・弗里斯的继承发展:科伦斯沿用 “天性”(Anlage)描述单个基因,德・弗里斯在达尔文 “泛生论” 基础上提出 “泛生子”(Pangene),首次为基因创造新名词。
3.约翰森的命名革新:1909 年,丹麦植物学家约翰森从 “泛生子”(Pangene)中剥离出 “Gene”,意为决定生物性状的细胞内物质,同时创造 “基因型”“表型” 等遗传学核心概念。
4.中文 “基因” 的完美翻译:1930 年,潘光旦将 “Gene” 译为 “基因”,取 “基本因子” 之意,兼顾音译(Gene 发音)与意译(遗传基本单位),超越原词含义。
二、刻在人类基因中的生存禁令
1.大脑功能的基因调控
-
神经信息处理禁令:DISC1 基因禁止大脑篡改感官输入信息,其失常会导致精神分裂症的幻觉幻听;COMT 基因调控多巴胺稳态,异常时可能引发双相情感障碍。
-
情绪与行为限制:5 - HTTLPR 等基因禁止长时间情绪消沉,CACNA1C 基因抑制躁狂发作,SHANK3 等基因阻止自闭行为,维持社交能力。
2.进化形成的生理保护机制
- 晕车反应的生存逻辑:平衡系统基因使大脑在运动感知与肌肉状态矛盾时触发呕吐,这是祖先避免有毒食物的进化策略,却在交通工具时代导致晕车。
- 梦境快速遗忘机制:基因设定梦境记忆快速消退,避免无序梦境干扰现实行为,如防止将梦中跳楼等危险行为误判为真实经验。
三、基因研究的双重意义
1.科学史的里程碑:从孟德尔的模糊描述到约翰森的精准命名,“基因” 概念的演进见证了遗传学从现象观察到分子机制的跨越。
2.进化生物学的活证据:基因禁令揭示自然选择如何通过 DNA 编码塑造人类行为,如平衡系统与梦境遗忘机制均为百万年进化的生存智慧结晶。
结语:基因既是承载遗传信息的分子密码,也是书写生命禁令的进化史册。从 “Gene” 的命名到 DNA 中镌刻的生存法则,人类对基因的认知始终贯穿科学探索与生存智慧的双重脉络,不断揭示生命本质的奥秘。
“基因”(Gene)一词是怎么来的? | 商周专栏
原创 商周 知识分子

图源:pixabay.com
导 读
✚ ● ○
基因所代表的物质在生命中至关重要,它的发现是科学史一个伟大的里程碑。“基因” 这个词的发明和翻译也堪称完美。
旅德免疫学学者、《知识分子》专栏作者商周,在本文介绍了基因(Gene)一词的由来。
撰文 | 商周
责编 | 陈晓雪
● ● ●
对科学名词的翻译的方式有两种,一种是意译(根据含义来翻译),另一种是音译(根据读音来翻译)。能在意译和音译上都达标则效果更佳,但这样的名词极少,一个难得的例子是 “基因”(Gene)。
把 “Gene” 翻译成基因,在含义和读音方面都达到了要求,相比其它广为人知的生物学名词(比如细胞(Cell)、器官(Organ)、组织(Tissue))的翻译,明显要更胜一筹。
那么,“基因”(Gene)这个词是如何来的呢?
1 孟德尔的 “天性”(Anlage)和 “因子”(Elemente)
词汇不是凭空出现的,只有当人们需要描述一个新鲜事物的时候,创造一个新的名词才成为必要。“基因” 这个名词的起源,也就是人类首次意识到基因这个物质存在的时候。第一个意识到基因这种物质存在的人,正是发现了遗传学法则的孟德尔,但他并没有为它去创造一个新的名词。
1854 年到 1863 年,孟德尔利用 22 种不同的豌豆品种进行杂交实验,发现豌豆不同的性状(比如种子颜色、形状,豆荚颜色、形状等)是由不同遗传物质控制的,这些遗传物质来自父母双方,而且来自父母双方的遗传物质在产生生殖细胞时会发生分离。现在我们知道,这些遗传物质就是基因。但在只有普通光学显微镜的十九世纪,人们对生命的认知还停留在细胞水平,虽然知道了细胞核的存在,但并不知道染色体,更不知道 DNA。
面对控制豌豆性状的神秘遗传物质,孟德尔在他的《植物杂交实验》论文里采用了两个不同的词来描述 [1]。
在论文结果部分的 “杂交种的生殖细胞” 单元,开头一段有如下描述:

图 1 孟德尔《植物杂交实验》论文截图 | 图源:biodiversitylibrary
“ …… 就经验而言,我们发现每一种情况下都证实,只有在卵细胞和受精花粉具有相同的天性(Anlage)时才能形成不变的后代,正如纯种植株的正常受精一样…….”
德语 “Anlage” 一词有七种含义(包括创造、投资、设施、装置、结构、天性、材料),在孟德尔上面的文字里,翻译成 “天性” 可能相对贴切一些。在这里,孟德尔用 “天性”(Anlage)这个词来描述豌豆花粉细胞和卵细胞里含有的遗传物质,因为这一段之前论文已经描述了单个和多个性状杂交的情况,这里的遗传物质并不是指单个基因,而是多个基因或者是整个生殖细胞里的所有遗传物质。
有趣的是,“天性”(Anlage)这个词孟德尔在整篇文章里只使用了这一次。等到文章的结语部分讨论控制性状的遗传物质的时候,他用了另外一个名词 “Elemente”,而且用了 10 次之多。Elemente 有三种不同的含义:基本成分、特质、因子。从孟德尔论文的语境来看,这里的 “Elemente” 翻译成 “因子” 更合适一些。

图 2 孟德尔《植物杂交实验》论文截图 | 图源:biodiversitylibrary.
而这里的 “因子”(Elemente)的具体含义,可以通过论文的这一段文字判断出来。
“对于那些后代存在变化的杂交种,我们也许可以假设,在卵细胞和花粉细胞的差异因子之间发生了某种协调,以至于作为杂交种基础的细胞的形成成为可能;但尽管如此,不同因子之间的平衡只是暂时的,并没有持续到杂交植物的整个生命中。由于植物的习性在整个植被期没有变化,我们必须进一步假设,只有当生殖细胞发育时,差异因子才有可能从强制结合中解放出来。在这些细胞的形成过程中,所有现有的因子都参与了一个完全自由和平等的分配,只有这样它们才会相互分离。这样一来,所产生的卵细胞和花粉的类型在数量上就和因子所可能形成的组合一样多。”
从这一段文字来看,孟德尔不仅谈到了来自父母的差异 “因子”(Elemente)的分离,也谈到了不同 **“因子”**的组合。所以,这里的 “因子” 指的就是单个的基因。而上面的 “天性”(Anlage)指的则是细胞内整个的遗传物质。正是因为这一微妙的差异,孟德尔选择了两个不同的单词进行描述。
尽管孟德尔用了这两个词对遗传物质在整体层面和单个基因层面进行描述,但因为其经典论文《植物杂交实验》长期被忽视,这两个词就更不可能走进人们的视野。直到 1900 年,荷兰植物学家胡戈 - 德弗里斯(Hugo de Vries)、德国植物学家卡尔 - 科伦斯(Carl Correns)以及瑞士植物学家埃里克・切尔马克(Erich Tschermark)分别在《德国植物协会通报》(Ber. der Deutschen Bot. Gesellsch.)上发表关于植物杂交的研究论文,各自独立地部分重现了孟德尔的发现 [2-4]。在这三个 “孟德尔的发现者”(注:学界对三人在这一主题上的贡献有争议,这里不仔细讨论)里,有两人也对因子(Elemente)这种神秘的物质进行了描述。
2 科伦斯的 “天性”(Analge)
科伦斯 1864 年出生在德国慕尼黑, 28 岁那年在德国图宾根大学获得植物学讲师职位,并在那里花了六年的时间进行植物杂交实验,重现了孟德尔的部分结果。在 “孟德尔的发现者” 的三人中,科伦斯对孟德尔的发现最为了解。他 1900 年发表在《德国植物协会通报》上的论文,标题就是《关于品种杂交后代行为的孟德尔法则》[3]。
在这篇论文里,科伦斯详细地讨论到控制性状的遗传物质。有趣的是,科伦斯用的词是 “天性”(Anlage), 而不是 “因子”(Elemente),而且整篇论文里一共提了 22 次。比如,在下面这段文字里:

图 3科伦斯《关于品种杂交后代行为的孟德尔法则》论文截图**|**图源:biodiversitylibrary
“为了解释这些事实,我们必须假设(就像孟德尔那样),在生殖核融合之后,一个性状,即隐性性状(在我们的例子中为绿色)的天性被另一个性状,即显性性状所抑制,因此所有的胚胎都是黄色。然而,虽然隐性性状的天性‘潜伏’ 着,但在生殖核的最终形成之前,两种性状的天性完全分离,所以一半的生殖核接受隐性天性,即绿色;另一半接受显性天性,即黄色……”
从文中的语境来看,科伦斯用 “天性” 描述的其实是控制性状的单个基因,而不是所有遗传物质的总和。所以,他虽然用的是 “天性” 这个词,但和孟德尔用的 “因子” 这个词一样,代表的都是基因。科伦斯之所以选择 “天性”,而没有使用 “因子”,一个可能的原因是受他的导师慕尼黑大学植物学家卡尔・威廉・冯・内格里(Carl Wilhelm von Nägeli)的影响。就在科伦斯进入慕尼黑大学的前一年(1884 年),内格里发表了他的巨著《生命进化的机械生理学理论》(Mechanisch-physiologische Theorie der Abstammungslehre),里面谈到遗传物质的时候,使用的就是 “天性”(Anlage)这一名词。但信奉融合遗传的内格里用 “天性”(Anlage)一词描述的不是基因,而是整体上的遗传物质。
无论是孟德尔,还是科伦斯,两人都清楚地意识到基因这种物质的存在,但并没有为它去创造一个新的名词,而是试着用已经存在的名词对其进行描述。
真正尝试为基因去创造新名词的,首先是德・弗里斯。
3 德・弗里斯的 “泛生子”(Pangene)
德・弗里斯 1848 年出生在荷兰,30 岁生日那天他获得了荷兰阿姆斯特丹大学的植物生理学教授职位,并在同年当选为荷兰科学和艺术学院的会员。1899 年,德・弗里斯写出了他的代表作之一《细胞内泛生论》(Intracellular Panenesis)一书。虽然是荷兰人,德・弗里斯的这本书是以当时科学界更为普及的德语出版的 ,出版社是位于德国耶拿的 GUSTAV FISCHER [5]。
“‘泛生论’(Pangenesis)一词包括两个希腊单词:Pan 和 Genesis,前者的意思是全部(泛),后者的意思是出生和起源(生)。这是达尔文 1868 年提出的一个有关遗传的理论,它的核心是融合遗传(Blending inheritance)。按照泛生论,生物体各部分的细胞都带有特定的自身繁殖的 ‘微芽’(后人也把微芽称微泛生子(Pangene)),这些 ‘微芽’ 可由各系统集中于生殖细胞,父母生殖细胞的 ‘微芽’ 会相互融合从而形成新的子代 ‘微芽’。和泛生论不同,由孟德尔开创的现代遗传学的核心是颗粒遗传(Particulate inheritance),即控制性状的基因是独立的单位,来自父母的两个等位基因并不会发生融合,在下一代形成生殖细胞时还会相互分离。”
德・弗里斯在 1898 年提出的 “细胞内泛生论” 则有些特别,一方面它依然是泛生论,另一方面它抛弃了融合遗传,提出了颗粒遗传的概念。正是因为提出了颗粒遗传这个概念,德・弗里斯以达尔文创造的 “泛生论”(Pangenesis)一词为基础,提出了 “泛生子”(Pangene)一词,并对这一概念进行了描述:

图 4 德・弗里斯的《细胞内泛生论》对 “泛生子”(Pangene)做了详细注解 | 图源:biodiversitylibrary
“…… 每个生殖细胞都必须潜在地包含构成相关物种性状的所有因素。因此,可见的遗传现象,都是隐藏在生命物质中的最小不可见粒子的特性的表现。事实上,为了能够解释所有的现象,人们必须为每个遗传属性假设特殊的粒子。我将这些单位称为泛生子(Pangene)。这些泛生子小得无法看见,但它们的化学分子的顺序完全不同,这些泛生子能够随着细胞分裂而增殖,并且可以分布到生物体所有或几乎所有的细胞中。它们要么是潜伏的,要么是活动的,但可以在这两种状态下繁殖……”
从上文可以看到,德・弗里斯所提到的 “泛生子” 其实就是基因。德・弗里斯提出的细胞内泛生论最有价值之处,就是提出了颗粒型遗传这一概念,否定了之前的融合性遗传。德・弗里斯能做到这一点,是因为在这之前进行了六七年的植物杂交实验,并且重现了孟德尔关于分离法则的发现。这让他意识到来自父母双方遗传物质并不会融合,而是依然会在产生生殖细胞时分离。
虽然 “泛生子” Pangene)是为基因创造的一个新名词,但这个词的前缀(Pan)用来描述基因并不合适,可以说有画蛇添足之嫌。1909 年,丹麦植物学家维尔海姆・路德维希・约翰森(Wilhelm Ludvig Johannsen )在 “Pangene” 一词的基础上,进一步提炼出了 “基因”(Gene)一词。
4 约翰森的 “基因”(Gene)
维尔海姆・路德维希・约翰森(Wilhelm Ludvig Johannsen)1857 年出生于丹麦的哥本哈根,1905 年获得哥本哈根大学的植物学教授职位。1909 年,他出版了自己的代表作《精确遗传学理论的要素》一书。和德・弗里斯的《细胞内泛生论》一样,约翰森《精确遗传学理论的要素》一书也是由德国耶拿的 GUSTAV FISCHER 出版社用德语发行 [6]。

图 5 约翰森的《精确遗传学理论的要素》一书的截图 | 图源:biodiversitylibrary
这本书由约翰森的一系列讲义组成,在其中的第八讲,他创造了 “基因” 一词。同时,约翰森还创造了 “基因型”(Genotype)、“表型”(Phenotype)、“纯合子”(Homozygote)、“杂合子”(Heterozygote)等今天常用的一系列遗传学术名词。
关于为什么要创造 “基因” 一词,约翰森在书中是这么说的:
“性细胞含有 ‘某种东西’,它决定着通过受精而产生的生物体的性状。这种 ‘东西’通常被称为 ‘天性’(Anlage),但这种说法相当含糊。达尔文提出的 ‘泛生子’(Pangene)一词,经常被用来代替 ‘天性’(Anlage)。然而, ‘泛生子’(Pangene)这个词的选择也并不令人满意,因为它是一个双重结构,包含了 ‘Pan’ 和 ‘Gene’两个词干。这里只需要考虑后者的意义,因此,从达尔文这个众所周知的词中分离出我们唯一感兴趣的最后一个音节 ‘Gene’,以便用它来取代糟糕的、模棱两可的 ‘天性’(Anlage)一词……”
在上面这段文字里,约翰森对基因是什么的描述比之前的任何一位学者都更清晰,即细胞里的 “某些东西”,能决定生物的性状。他也指出,之前用来描述基因的词 “天性”(Anlage)以及 “泛生子”(Pangene)都有不足,前者太模糊,后者前面带了多余的修饰词。所以,约翰森把 “Gene” 从 “Pangene” 中剥离了出来。
接下来,约翰森还进一步说明了使用 “基因”(Gene)这个词的优势:
“……‘基因’ 这个短词有很多优点,因为它可以很容易地与其他名称组合。如果我们想到由某个 ‘基因’ 决定的属性(比如财富),我们就可以很容易地说 ‘财富的 基因’,而不需要使用 ‘决定财富的 基因’ 这样更繁琐的短语。”
不知为什么,约翰森在提到前人描述基因所用的词汇时,关于 “泛生子” 一词的发明,只提到了达尔文,而没有谈到德・弗里斯。还有,约翰森也没有提到孟德尔首次用的 “因子”(Elemente)这个词。不过,约翰森没有忘记把发现基因这一里程碑式的发现归功于孟德尔:
“…… ‘基因’ 的性质,目前还没有足够充分的依据。然而,这对遗传研究的有效性没有任何影响;只要确定存在这样的 ‘基因’ 就足够了。它的发现是格雷戈尔・孟德尔开展的植物杂交实验研究的最重要成就之一……”
在 1909 年 “基因” 这个词被创造出来的时候,正如上文约翰森提到的,人们对基因的自然属性还并不了解,只是知道它的存在,知道它是生物性状的决定者。但这已经足够了,因为这开辟了一个全新而且重要的研究领域。后来,人们知道了基因是染色体上的一部分;再后来,人们知道了基因是编码一段多肽的 DNA 片段……
5 从 “Gene” 到 “基因”
把 “Gene” 翻译成中文 “基因”,不仅同时做到了意译和音译,而且提高这个单词在含义上的准确性。就像上面提到的,“Gene” 这个词来源于希腊语,本来的意思是 “出生” 和 “起源”,这和 “决定生物性状的遗传物质” 的本意不太一致。但当把它翻译成 “基因”(基本因子)后,就和本意靠近了很多,因为 “基本因子” 同时涵盖了孟德尔的 “因子”(Elemente)、科伦斯的 “天性”(Anlage)、约翰森的 “基因”(Gene)。从某种角度上来说,将 “Gene” 翻译成 “基因” 是对原词的一个提升和超越。
那么,是谁做了这样一个完美的翻译呢?
根据加拿大曼尼托巴大学医学院谢永久教授的考证 [7],目前能查到的中文资料里,最早翻译 “Gene” 为 “基因” 一词的是潘光旦先生,他在 1930 年发表的《文化的生物学观》一文中写道 [8]:
“关于遗传这一点,我们不预备多说。遗传的几条原则,什么韦思曼的精质绵续与精质比较独立说、孟特尔的三律、跟了韦氏的理论而发生的新达尔文主义或后天习得性不遗传说、杜勿黎的突变说、约杭生与摩尔更的 ‘基因’ 遗传说 —— 是大多数生物学家已认为有效,而且在生物学教本中已经数见不鲜的。”
潘光旦在 1930 年(可能更早)首次将 “Gene” 翻译成 “基因” 并非偶然,1922 年 23 岁的他留学美国,并于 1926 年在哥伦比亚大学获得生物学学位,那里的教授里就有著名的遗传学大师摩尔根。虽然潘光旦后来成为了一名出色的社会学家,但他早期从事过一些优生学的研究,比如在 1923 年就发表了《优生学在中国》(Eugenics and China)的英文论文,并在随后将优生学引入到中文世界。或许正是因为他的自然和社会科学的双重背景,成就了 “基因” 的完美翻译。
参考文献:
- Mendel, G., 1866 Versuch e über Pflanzen-Hybriden. Verh. naturf. Ver. Brünn 4: 3–47.
- De Vries, H. Das Spaltungsgesetz der Bastarde. Ber. der Deutschen Bot. Gesellsch. 18 (3): 83, 1900.
- Correns C. G. Mendel’s Regel über das Verhalten der Nachkommenschaft der Rassenbastarde. Ber. der Deutschen Bot. Gesellsch., 18 (4): 158-168, 1900.
- Tschermak, E. Über Künstliche Kreuzung bei Pisum sativum. Berichte der Deutsche Botanischen Gesellschaft 18: 232-239, 1900.
- de Vries, Hugo. 1889Intracellular Pangenesis. Gustav Fischer, Jena.
- Johannsen, W., 1909 Elemente der exakten Erblichkeitslehre. Gustav Fischer, Jena.
- http://home.cc.umanitoba.ca/~xiej/genetranslation.pdf
- 《潘光旦文集》第二卷,潘乃穆,潘乃和 编, 北京 - 北京大学出版社, 1994 年 10 月,ISBN 7-301-02571-8, 318-319 页.
制版编辑 | 卢卡斯
知识分子 The Intellectual
刻在人类基因中的禁令有哪些?
知乎日报 2024年09月05日 21:00 北京
几万年来,人类在自然环境中生存发展,形成了一些基因禁令。这些禁令与人类的生存发展有着紧密的联系,无论是对健康身体的保护,还是对生命安全的保护,都具有重要的作用。刻在人类基因中的禁令有哪些?一起来看看答主们的回答吧。
刻在人类基因中的禁令有哪些?
| 答主:SACE
近现代的心理学研究是进入了人类大脑领域的,如果你认为那些基因阻止人类有意或无意的行为就算是禁令的话,那与脑部相关的基因每一条都写满了「禁止」两个字。
第一组:由 DISC1、COMT、NRG1 等基因操控
-
禁止篡改器官输入的外界信息
-
禁止自杀
-
禁止无意义恐慌
-
禁止持续性焦虑
第二组:由 5-HTTLPR、BDNF、SERT 等基因操控
-
禁止长时间的情绪消沉
-
禁止长时间地停止行动
第三组:由 CACNA1C、ANK3 等基因操控
-
禁止躁狂
-
禁止激素调整过于快速
第四组:由 SHANK3、NLGN3、NLGN4 等基因操控
-
禁止自闭
-
禁止拒绝感受情感
当这些基因不能正常表达时,其对应形成的蛋白质将无法正常工作,其结果就是人类整体出现非正常运行的情况,以上基因阻止了人类大脑和其他器官的很多行为。 比如说,如果没有 DISC1 基因或其失常,你的大脑可以不必接收眼睛传来的视觉信息,耳朵传来的听觉信息,而是自己自行编造外界信息,说白了就是:精神分裂症,幻觉,幻听,幻视。
如果没有 CACNA1C 基因或其失常,你的大脑可以怎么舒服怎么来的向其他部位排放调节激素,导致人类情绪变化起伏不定,波动极大,典型的就是双相情感障碍,其易感人群多能发现相应基因的异常。
下面具体介绍其中一些基因:
DISC1 是一种胞质支架蛋白,具有四个预测的卷曲螺旋结构域;翻译后修饰理论上可以包括磷酸化、苏木化、多聚化和蛋白水解成活性片段。
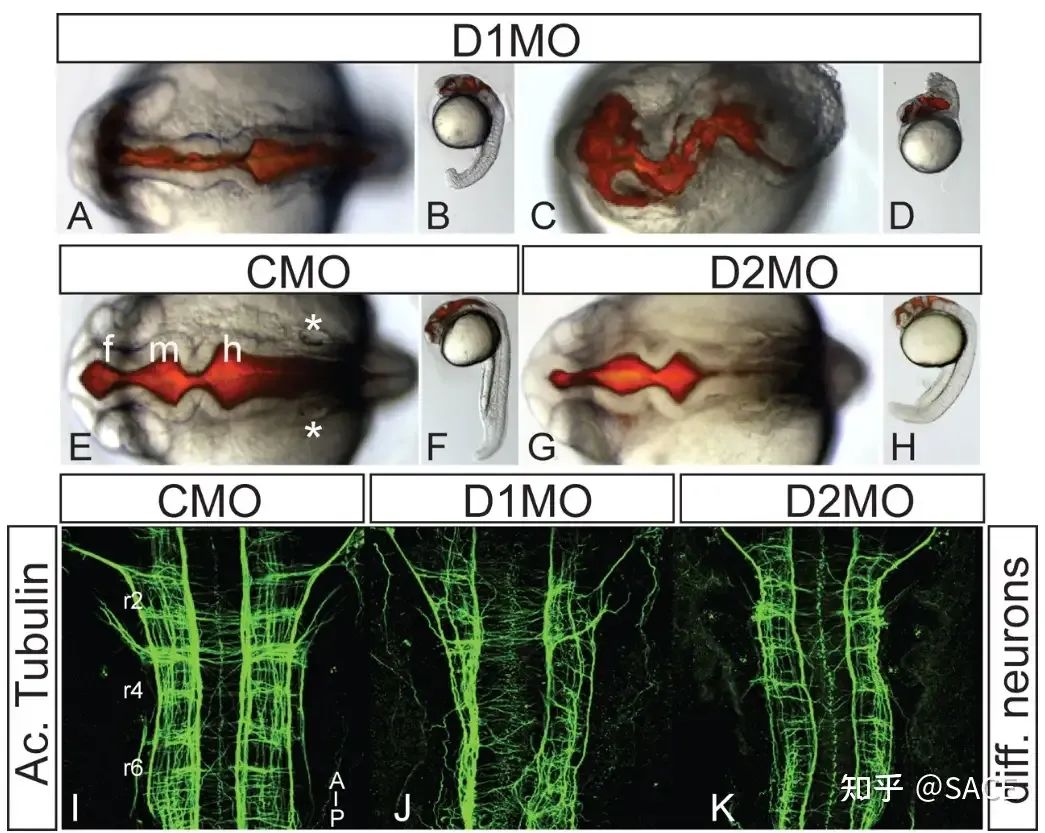
**斑马鱼和哺乳动物的 DISC1 基因中存在保守结构域。**斑马鱼 DISC1 在发育中的中枢神经系统中以母体表达,合子表达最强。为了测试 DISC1 的胚胎功能,研究员通过注射吗啉代修饰的反义寡核苷酸(MO)靶向剪接位点来降低合子基因功能。描述为 D1MO 的 DISC1 吗啉代针对外显子 1 和内含子 1/2 之间的剪接供体位点,并消除所有正常的 DISC1 mRNA。注射 D1MO 的胚胎表现出非常强的表型,早期体细胞发生很明显,包括前脑形成缺陷和整个大脑轴突生长的巨大失败(见图)。相比之下,第二个 DISC1 靶向吗啉代(D2MO)导致 DISC-1 蛋白在外显子 8 和 9 之间发生异常剪接和截断,但表型比 D1 严重得多。这种截断概括了在与精神分裂症相关的人类调查中看到的情况相同。这种截短蛋白在野生型胚胎大脑中的表达导致胚胎具有与用 D2MO 处理的胚胎相似的表型,表明它可能干扰正常的 DISC-1 功能。
COMT 是一个 S- 腺苷甲硫氨酸依赖性甲基转移酶,可甲基化儿茶酚底物,特别是儿茶酚胺和儿茶酚雌激素。 它以两种形式存在,可溶性(S-COMT)和膜结合(MB-COMT),源自不同的翻译起始位点。儿茶酚-O-甲基转移酶(COMT)是精神疾病中研究最深入的基因之一 ,它调节突触中神经递质多巴胺的稳态水平。该基因位于染色体 22q11.21 中,该区域被多项研究与精神分裂症和双相情感障碍相关联。
| 答主:真狗熊
人类的祖先当年在树上生活过,所以有很强的平衡能力,能够感受到颠簸和旋转。
强力的立体视觉配合上强力的平衡能力,可以让人类祖先在树上来去自如,简直完美。
当然,这么精密的平衡系统也有点小缺陷,那就是容易出问题。
我们的脑袋无论出什么毛病,最常见的症状就是头晕。
而对于我们的祖先来说,神经系统出问题最常见的原因是什么呢?
就是乱吃东西,吃到了有毒的食物。
所以我们的大脑长久以来形成了这么一套行为模式:
如果你身体感受到的运动状态和你实际的运动行为不一致。
那说明你肯定是吃了有毒的东西,影响了最敏感的平衡感知神经。
这时候刻在你基因里的行为模式就会发出指令:呕吐,赶快吧有毒的东西吐出来。
这个模式一直运行的很好,直到人类发明了交通工具…….
当你坐在汽车上,耳朵感受到身体在加速减速左转右转。但是你的肌肉却是放松状态,完全没有运动。
大脑一分析:坏了,我中毒了!赶快发出眩晕信号,开始吐!
| 答主:峡谷职业养爹人
有一个很少被提及的点,那就是——「梦境的快速遗忘」。
只要你稍微留心一下,你就会发现,无论是什么人种,什么年龄,什么性别,能够记住梦境的人类占比极少。 大部分人类在醒来后就会快速遗忘掉梦境中的记忆,哪怕你醒来后当时觉得记住了,但只要你不拿纸笔记录很多,几小时后就会模糊梦境中的记忆。而相对的,你昨天打游戏的八连跪反而能念念不忘。
**究其根本还是保证人类的生存。因为梦境是弱逻辑的,是无序混乱的,如果让这样的记忆残留,很容易影响现实中的行为。**举例来说,在梦中你能飞天跳楼,但现实中你只要这样玩就是个死。如果你记忆中梦境跳楼之类的行为做多了,一旦在现实中误判,你就得寄。所以我们的基因进化成梦境记忆快速遗忘的技能。它可能让我们的主观记忆少了一大截,但却能让我们活的更久。
via:
-
“基因”(Gene)一词是怎么来的? | 商周专栏 原创 商周 知识分子 2022 年 01 月 24 日 07:59
https://mp.weixin.qq.com/s/ZW_vwj_566O_hkxEevKhGg -
刻在人类基因中的禁令有哪些?知乎日报 2024年09月05日 21:00 北京
https://mp.weixin.qq.com/s/2G6e9N2HNwjB3aiMPtCQtQ

















 1849
1849

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









