









 研究人员通过第一性原理DFT计算,探讨了过渡金属掺杂对超薄PdM双金属材料在氧还原反应中的影响。他们发现,掺杂改变了Pd位点活性和稳定性,优选出PdTa、PdHf、PdZr和PdNb等催化剂,这些催化剂具有较低的过电位和较高的稳定性,为ORR电催化提供了新的设计策略。
研究人员通过第一性原理DFT计算,探讨了过渡金属掺杂对超薄PdM双金属材料在氧还原反应中的影响。他们发现,掺杂改变了Pd位点活性和稳定性,优选出PdTa、PdHf、PdZr和PdNb等催化剂,这些催化剂具有较低的过电位和较高的稳定性,为ORR电催化提供了新的设计策略。

研究背景
作为非铂电催化剂的替代品,双金属钯基催化剂在电催化反应中发挥着至关重要的作用。而将过渡金属(M)掺杂到超薄钯纳米片中是一种有效策略,其可以调节表面钯位点的反应活性和耐久性。近日,重庆大学唐青等人对2D超薄PdM双金属材料上的氧还原反应(ORR)活性和稳定性进行了深入研究。
计算方法
作者使用VASP 5.4.4版进行了第一性原理密度泛函理论(DFT)计算,并利用广义梯度近似(GGA)的投影增广波赝势(PAW)和Perdew–Burke–Ernzerhof(PBE)交换相关泛函来描述核心电子和价电子。作者使用能量截断为400 eV的平面波基组,并用DFT+D3对范德瓦尔斯(VDW)力进行了校正。作者采用四层(4×4)超胞fcc板对纯钯(111)表面进行模拟,并通过用TMs取代中间层的钯原子来构建钯M合金。在PdM模型中,所有的原子层和吸附的物种都保持弛豫。在DFT计算中,作者对布里渊区采用(3×3×1)Monkhorst–Pack网格采样。为了避免周期性相互作用,作者在z方向上设置了20Å的真空层。在几何优化过程中,能量收敛标准设置为10−4 eV,力的收敛标准设置为0.01 eVÅ−1,并采用泊松-玻尔兹曼隐式溶化模型来考虑水溶剂化的影响。作者采用CI-NEB方法来搜索O2离解的过渡态(TS)。
结果与讨论
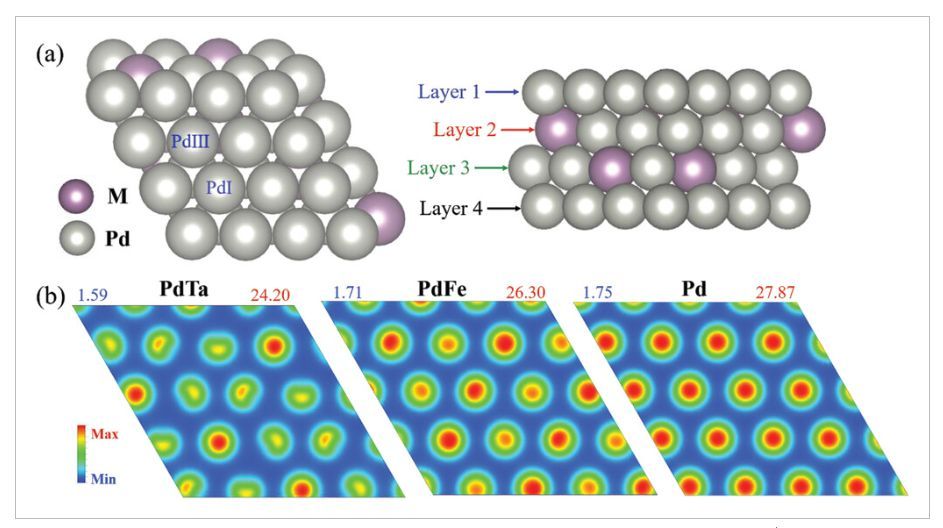
图1 模型结构和SF(r)
如图1a所示,在PdM(M=3d、4d和5d过渡金属)双金属模型中,第2层和第3层中的M原子比例为25%,而在第1层和第4层中为0%。为了验证该模型合理性,作者随机测试了三种结构,其中两种是位于表面上的50%M原子(表示为PdM0.5),第三种是所有M原子位于表面上(表示为PdM1)。计算结果表明,对于大多数PdM双金属(除了PdAg、PdHg和PdAu),所有M原子嵌入PdM双合金中的结构更稳定。为了定量描述各种PdM双金属中的表面Pd反应活性,作者计算了SF(r)(图1b),与纯Pd表面相比,PdM双金属表面Pd反应活性变弱。对于早期过渡金属(例如PdTa、PdMo),表面反应活性降低幅度较大,而对于晚期过渡金属(如PdFe、PdPt),表面活性降低幅度较小。此外,与次表面M掺杂直接结合的表面PdIII原子具有降低的反应活性,而与次表面Pd原子结合的表面PdI原子的反应活性几乎保持不变。从分子轨道(MO)的角度来看,早期过渡金属的价电子通常小于Pd(4d10)的价电子,导致Pd–M键态的占有率更高,金属之间的键合更强,从而削弱了表面PdIII原子的反应活性。
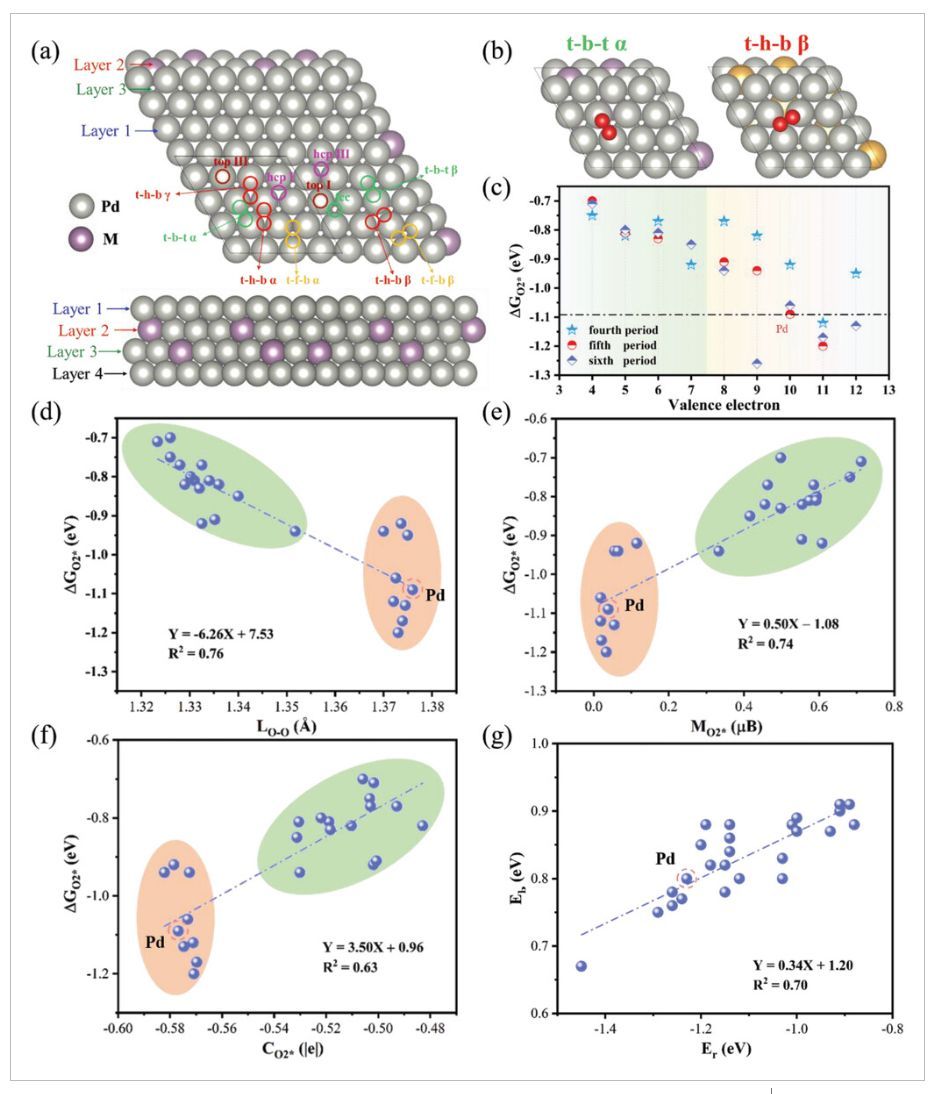
图2 吸附位点、吸附自由能与价电子、O-O键长、自旋磁矩、Bader电荷的关系、O2解离反应能与结合能的关系
如图2a所示,作者考虑了O2*(如t-b-tα、t-h-bβ)和O*(如fcc、hcp I)在PdM双金属上的吸附位点。强吸附O2的O–O键轴几乎平行于PdM表面(图2b)。而对于早期过渡金属掺杂的Pd(如PdTi、PdW),O2更倾向于吸附在t-b-tα位点。对于大多数后过渡金属掺杂的Pd(如PdAu、PdRh),O2更倾向于吸附在t-h-bβ位点,而对于一些价电子较少的后过渡金属(如PdFe、PdIr),t-b-tα位点最稳定。从图2c中可以看出,随着金属价电子的增加,O2吸附增强,并且大多数PdM双金属的吸附强度弱于纯Pd(111)。通过接受来自Pd表面的自旋向下电子,将磁矩从O2转移到金属,由自旋向上电子占据的O2反键π*轨道(即LUMO)被进一步填充。因此,吸附O2的键长(LO–O)、磁矩(MO2*)和Bader电荷(CO2*)与O2活化有关,这些因素与O2吸附自由能(ΔG)之间的相关性如图2d–f所示。对于早期或部分晚期过渡金属PdM双金属(绿色区域),O–O键长分布在1.32和1.35Å之间,而对于大多数晚期过渡金属PdM系统(橙色区域),其接近1.37Å。LO–O和ΔGO2*之间的决定系数R2为0.76(图2d),LO–O参数可以更好地描述早期过渡金属PdM双金属的吸附性能(绿色圆圈)。同样,作者还分析了ΔGO2*与吸附O2的磁矩(MO2*)和Bader电荷(CO2*)之间的相关性。根据图2e,f,ΔGO2*和MO2*或CO2*之间的决定系数R2为0.74和0.63,表明它们也是O2活化的良好描述符。此外,作者进一步探索了这些描述符之间的相关性,发现LO–O、MO2*和CO2*之间存在线性相关性,并分别具有0.96、0.89和0.84的高R2,表明吸附O2的几何和电子性质高度相关。
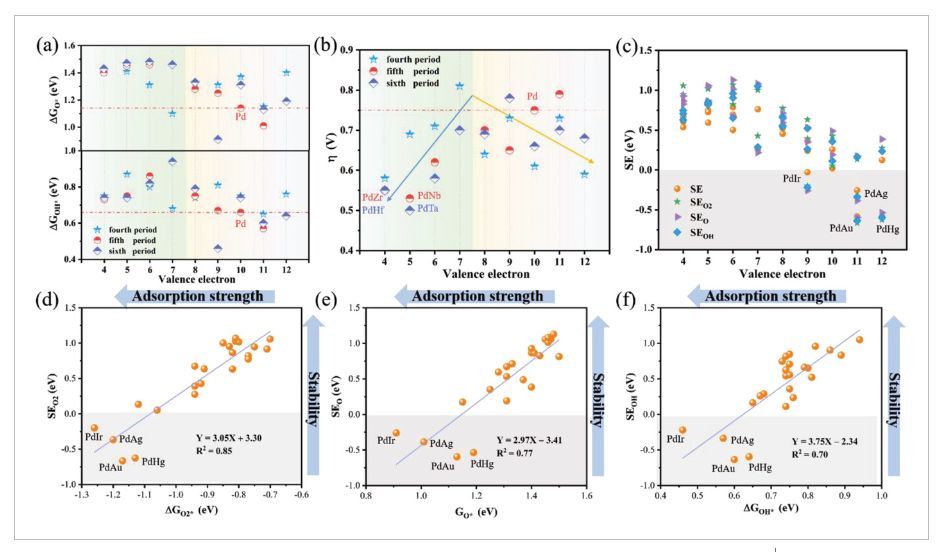
图3 ΔGO*和ΔGOH*、过电势、偏析能与价电子关系、偏析能与吸附自由能关系
如图3a所示,O*和OH*中间体在大多数PdM双金属上的吸附变得比在Pd(111)上的吸附更弱(红色虚线)。从图3b中可以看出,24种PdM催化剂上的过电势(η)与M掺杂剂的价电子数呈反V型关系。在早期过渡金属PdM双金属(绿色区域)的情况下,η随着M价电子的增加而逐渐增加。相反,对于后过渡金属PdM双金属(黄色区域),η呈下降趋势。与纯Pd(111)(η=0.75V,红色虚线)相比,大多数PdM双金属的η值要低得多,这表明金属合金化可以大大提高表面Pd位点的ORR活性。其中,PdTa、PdHf、PdZr和PdNb可以极大地提高ORR活性,并且其过电位最低(η≤0.55V)。除了电催化活性外,作者发现PdM双金属催化剂的结构稳定性也很重要。PdM双金属的结构稳定性可以通过计算偏析能(SE)来评估,其中偏析能为将M原子从Pd的亚表面移动到Pd的顶表面所需的能量。因此,更正的SE意味着PdM合金结构具有更高的稳定性。否则,负SE意味着M可能会迁移到表面,并导致PdM结构热力学不稳定。图3c是PdM双金属和具有不同ORR中间体(O2*、O*和OH*)的吸附PdM双合金的SE值。可以清楚地看到,PdAg、PdIr、PdAu和PdHg双金属具有较差的稳定性(灰色区域)。M掺杂剂在O2*、O*和OH*吸附的PdM双金属中的偏析行为可以通过O2*(ΔGO2*)、O*(ΔGO*)和OH*(ΔGOH*)的吸附自由能很好地预测,它们的相关系数分别为0.85(图3d)、0.77(图3e)和0.70(图3f)。进而表明,对O2*、O*或OH*的吸附越强,PdM双金属框架的结构偏析和稳定性就越差。考虑到ORR活性和催化剂稳定性,PdZr、PdNb、PdHf和PdTa是极具潜力的催化剂,其具有较低的过电位(η≤0.55V)和较高的稳定性(SE值>0.5)。
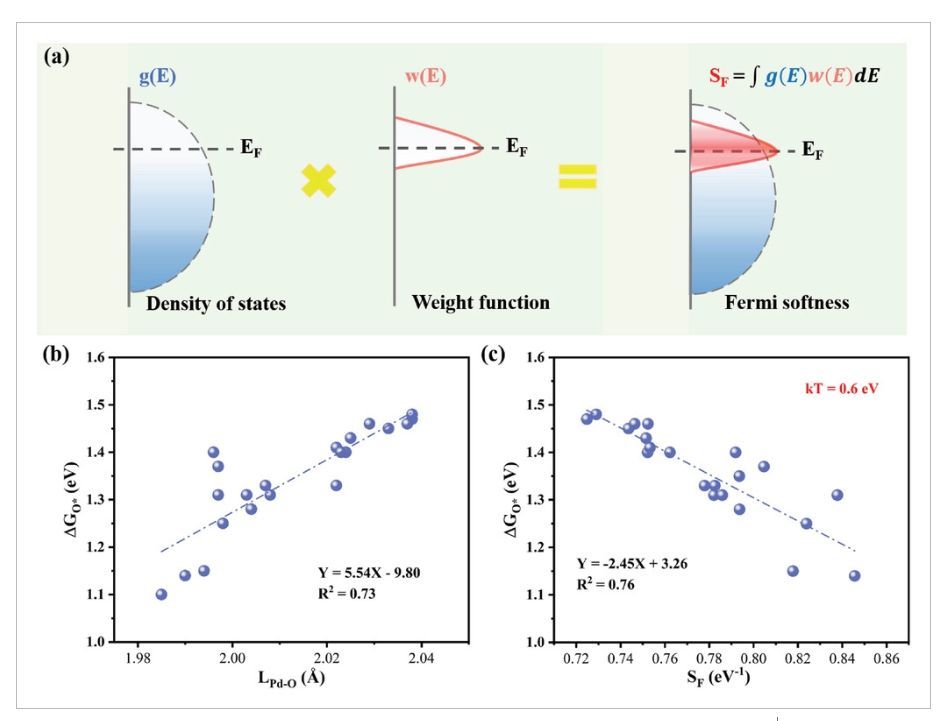
图4 SF、ΔGO*与LPd–O、SF的关系
在ORR中,不同的电催化活性源于关键吸附物种在不同催化剂上的不同结合强度。O*吸附自由能(ΔGO*)在合金体系的活性中起着重要作用。在PdM双金属中,ΔGO*的范围为0.91至1.48eV。就几何结构而言,O*吸附的PdM中的Pd−O键长(LPd−O,Å)与O*结合强度有关。从图4b中可以看出,LPd−O和ΔGO*之间的决定系数R2为0.73,表明Pd表面的O*吸附强度可以很好地用Pd–O键长来描述。就电子结构而言,O*的电荷状态可以反映Pd和O*之间的电子转移。SF的定义如图4a所示,其中更大的SF值意味着费米能级附近占据态的分布更高,因此对应于与O*的更强的结合强度,反之亦然。对于PdM双金属纳米合金,0.72至0.75eV的最佳SF范围将对应于相对高的ORR活性(图4c)。
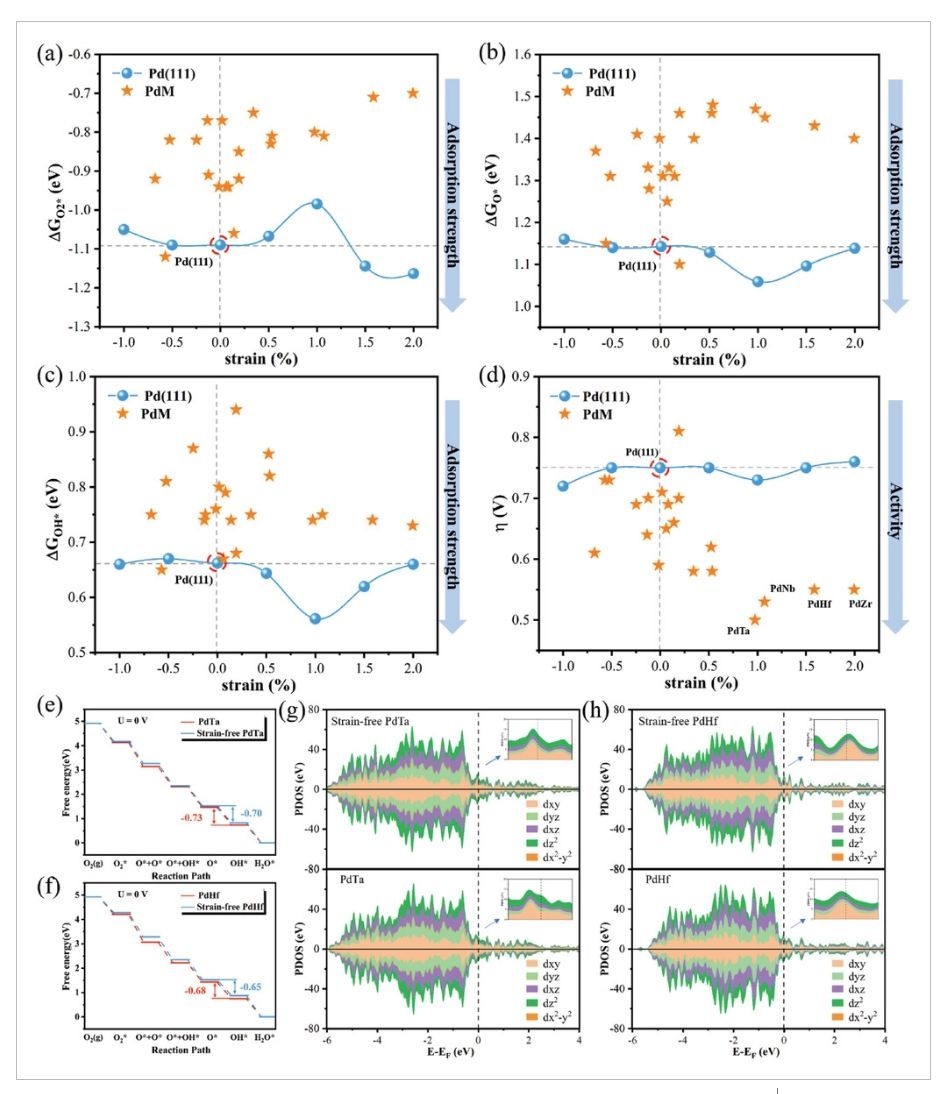
图5 应变与吸附自由能、过电位的关系、反应势能面和PDOS
O2*、O*和OH*的吸附强度,以及ORR过电位与应变的关系图如图5a–d所示,过电位仅从未应变的Pt(111)中的0.75 V略微降低到−1%压缩应变下的0.72 V和1%拉伸应变下的0.73 V。Pd(111)中单独应变引起的吸附强度和过电位的变化要比PdM双金属中的变化小得多(如图5a–d中的星形数据点所示)。这表明晶格应变在PdM双金属的活性增强中起到的作用可以忽略不计。相反,大多数PdM双金属中表面ORR活性的调控主要由杂金属掺杂的配体效应贡献。为了研究配体效应,作者将所有PdM双金属的晶格参数都固定为纯Pd(111)金属的晶格参数,从而使Pd表面的电子或化学性质的任何变化都由来自亚表面金属原子的配体效应引起。作者研究了PdTa、PdHf、PdZr和PdNb中的配体效应(图5e,f),从O*到OH*的速率决定步骤中的反应自由能在弛豫和晶格固定的PdTa(-0.73 eV vs-0.70 eV)和PdHf(-0.68 eV vs−0.65 eV)中非常接近。并且在PdNb和PdZr中也观察到类似的现象,进而表明金属诱导的配体效应是影响PdM双金属活性的主要因素。作者通过对电子态密度的进一步分析表明,在弛豫和晶格固定的PdTa(图5g)和PdHf(图5h)中,费米能级附近Pd-d电子态的分布和占据相似,即费米能级周围的金属电子态主要由dz2轨道贡献。因此,可以确定PdM双金属中表面Pd的电子性质和化学活性主要受到来自亚表面金属掺杂的配体效应的影响。
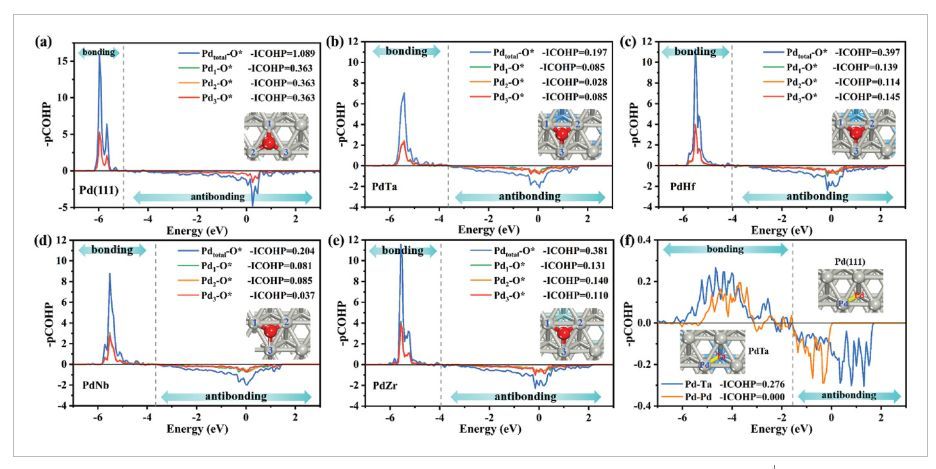
图6 COHP分析
作者计算了Pd(111)、PdTa、PdHf、PdNb和PdZr中Pd−O键的投影晶体轨道汉密尔顿布居(pCOHP)和积分COHP(ICOHP)(图6a–e),以确定O*和Pd活性中心之间的键合相互作用。在图6中,纯Pd(111)中的表面Pd表现出最强的Pd−O键合,−ICOHP为1.089 eV,其次是PdHf(0.397 eV)、PdZr(0.381 eV),PdNb(0.204 eV)和PdTa(0.197 eV)。这种削弱的Pd−O键与Pd−O键长(LPd−O)的变化趋势一致:Pd(1.990Å)<PdHf(2.025Å)≈PdZr(2.023Å)<PdNb(2.033Å)<PdTa(2.038Å)。此外,作者还对PdTa双金属中的Pd−Ta键进行了COHP分析(图6f)。PdTa中的表面Pd和亚表面Ta原子之间的键比纯Pd(111)中的Pd−Pd键强得多,如Pd−Ta键的–ICOHP值(0.276 eV)高于Pd−Pd键(0.00 eV)所示,这是d带中心下移(PdTa中为−2.00 eV,Pd(11)中为−1.48 eV)和表面Pd位点吸附减弱的根本原因。特别是,PdTa双金属中较弱的Pd−O键与对Pd−O键的键贡献提供了有效平衡,并导致ORR活性的显著增加。
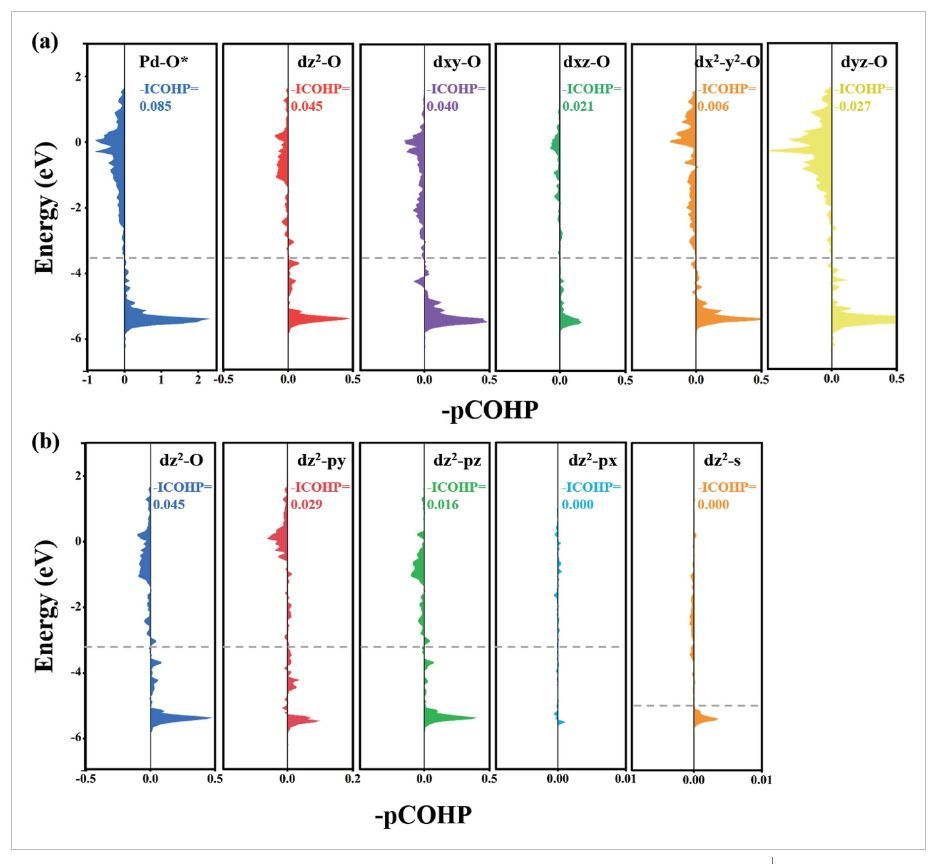
图7 -pCOHP分析
为了更深入地理解轨道相互作用,作者分析了PdTa双金属中活性Pd原子和O*吸附质的不同价轨道(dxy、dyz、dz2、dxz和dx2-y2)之间的成键和反键相互作用(图7)。Pd−O键主要由Pd-dz2轨道(-ICOHP=0.045 eV)贡献,其次是dxy(-ICOHP=0.040 eV;图7a)。此外,EF处的小峰值主要来自dz2-py相互作用(图7b)。根据上述pCOHP分析,在PdM双金属中引入亚表面杂金属原子对于调节活性Pd位点的dz2占据和防止氧中间体的过度结合以及导致ORR活性的提高至关重要。
结论与展望
M掺杂可以通过改变SF(r)来极大地改变Pd位点的反应活性。所有PdM双金属都遵循解离4e−途径,经过系统筛选,作者确定了几种有效的替代催化剂(PdTa、PdHf、PdZr和PdNb),其ORR过电位远低于纯Pd(111)金属。Pd–O键长和表面Pd原子的SF是O*关键中间体吸附的有效描述符。杂金属诱导的配体效应对活性的提高起着关键作用,它通过Pd−M轨道杂化改变了Pd的电子性质和表面反应活性,并导致Pd 3dz2和O*2p轨道之间的键填充下降。该工作为双金属钯基纳米合金在ORR和其他电催化反应中的未来实验实现提供了有用的指导。
文献信息
Yuping Chen et.al Computational Insights and Design of Promising Ultrathin PdM Bimetallenes for Oxygen Reduction Electrocatalysis Small Methods 2023
htt-ps://doi.org/10.1002/smtd.202300276

















 3331
3331

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









