






双重测序技术已广泛应用于循环肿瘤脱氧核糖核酸(DNA)低频突变的检测,但如何确定测序深度等实验参数以保证低频突变的稳定检测仍是亟待解决的问题。双链测序的突变检测规则不仅限制了突变模板的数量,还限制了突变模板的每条正向链和反向链对应的突变支持读数的数量。为解决这一问题,我们提出了双工测序中稳定检测低频突变的深度估计模型(DELFMUT),该模型使用零截断负二项分布来模拟模板和读数之间的身份对应关系和数量关系,而不考虑碱基组成的序列。DELFMUT 的结果得到了真实双工测序数据的验证。在已知突变频率和突变检测规则的情况下,DELFMUT可以推荐DNA输入和测序深度的组合,保证突变的稳定检测,在指导双工测序技术的实验参数设置方面具有重要的应用价值。
导言
无细胞脱氧核糖核酸(DNA)(cfDNA)是指细胞释放到体液中的游离 DNA [1-5]。通常,癌症患者血液中的 cfDNA 含量高于正常人 [6-8]。基于血浆 cfDNA 的突变检测已广泛应用于癌症的早期检测、诊断、预后和治疗后监测[9-18]。由于在大多数情况下,来自癌细胞的循环肿瘤 DNA(ctDNA)在 cfDNA 中的比例较低,通常小于 1% [19,20],因此在传统的高通量测序技术中很难将真正的低频突变与测序错误区分开来 [21-23]。在双链测序技术中使用了唯一分子识别码(UMI),首先将具有相同 UMI 的读数分成族/簇,构建单链共识序列(SSCS),然后将具有互补 UMI 的单链共识序列统一为双链共识序列(DCS)[24, 25]。在 DCS 中持续出现的突变被鉴定为真正的突变,从而能够与聚合酶链反应(PCR)和测序错误区分开来,并将理论背景错误率降低到 1/109 [24]。虽然双工测序解决了低频突变检测的技术问题,但要保证在多次实验中重复稳定地检测到低频突变仍是一个挑战[26]。提高测序深度可在一定程度上解决这一问题,而变异等位基因频率(VAF)越低,稳定检测突变所需的测序深度就越高。因此,必须确定测序的最低深度要求,以确保临床突变检测的准确性[26],而测序深度估计方法的不一致会限制临床实践的应用和推广[27]。
估计测序深度的常用算法是在读数或 DNA 模板水平上使用二项分布 [26]。将测序深度 D(总读数/总模板数)和突变频率 VAF 分别作为二项分布中的实验总数和成功概率,突变读数或模板数 X 服从 Binom (D, VAF) 分布。如果至少检测到 k 个突变读数或模板,则认为检测到了突变,突变的检测概率变为 P (X ≥ k)。反之,假设已知突变的检测概率 P (X ≥ k) 为 95%,则相应的实验总数 D 正是确保稳定检测到突变所需的测序深度。上述使用二项分布的测序深度估算仅在读数或模板层面执行。然而,在根据双工测序数据断言检测到突变时,突变模板的数量和与突变模板的每个正向链和反向链相对应的突变支持读数的数量(也称为族/簇大小)都受到限制。因此,设计一种适合双链测序的测序深度估计方法对于确保稳定检测低频突变至关重要。虽然家系大小为 3 或以上的簇通常被认为是可信的[24, 25],但 Gundula Povysil 等人发现,即使大小为 1-2 的簇也可能包含真正的突变,而且通过分析这些小簇可以检索到一些突变[28]。由于双链测序数据中只有部分读数被组装成 DCS,如果只保留这些双链模板,会丢失很多读数,因此单链模板也可纳入后续的突变检测过程[28]。双链测序技术的深度估计更多是在稀释样本的真实测序数据上进行的。具体来说,适当的测序深度是通过向下取样到不同深度并综合考虑突变检测的灵敏度和特异性来确定的[29]。
现有的基于模型的测序深度方法使用简单的二项分布,但由于没有考虑读数的聚类过程,因此不太适合双工测序技术。因此,我们提出了一种在双工测序中稳定检测低频突变的深度估计模型(DELFMUT)。DELFMUT 将输入的 DNA 质量(或饱和测序状态下的 DNA 模板数量)、VAF 和突变检测规则作为输入,在多个预设测序深度下输出突变检测频率。此外,我们还利用真实的双工测序数据验证了 DELFMUT 的结果。除了量化测序深度与检测频率之间的关系,DELFMUT 还能根据 VAF 和突变检测规则,推荐用于稳定检测低频突变的 DNA 输入和测序深度组合。DELFMUT通过指导双工测序的实验参数设置,实现了低频突变的重复和稳定检测,这将有助于cfDNA突变检测在癌症筛查和治疗中的临床应用和推广。
材料与方法
DELFMUT 概述
对于双工测序,我们将样本的饱和测序状态定义为:随着测序深度(测序读数的数量)的增加,检测到的 DNA 模板/片段的数量(也称为重复深度)在达到一定值后保持稳定。在饱和测序状态下,血浆样本的重复深度主要由 DNA 输入量和测序技术的捕获效率决定。除了样本水平的重复深度外,在饱和测序状态下,覆盖特定基因组位点的重复深度、双链模板比例以及读数水平和模板水平的链偏倚也趋于稳定,而在不饱和状态下,它们的数值变化很大。因此,为了得到不同测序深度下不同VAFs的突变检测概率,我们首先模拟饱和状态下单个基因座对应的模板和读数,以及带有突变的模板和读数(图1A),然后对这些读数进行多次下采样到指定的测序深度,以模拟该基因座在不同测序深度下的状态(图1B)。最后,使用大量模拟数据集的突变检测频率平均值作为检测概率的估计值。在 DELFMUT 中,只对模板和匹配读数之间的身份对应关系和数量关系进行建模,无需生成真实的碱基序列。根据 DELFMUT 的输出结果,在预先确定 DNA 输入量和突变检测规则的情况下,我们可以得到预设测序深度与不同 VAF 的突变检测概率之间的关系曲线。此外,为确保突变的稳定检测,当突变频率和突变检测规则预先确定时,DELFMUT 可推荐双工测序的 DNA 输入量和测序深度组合。
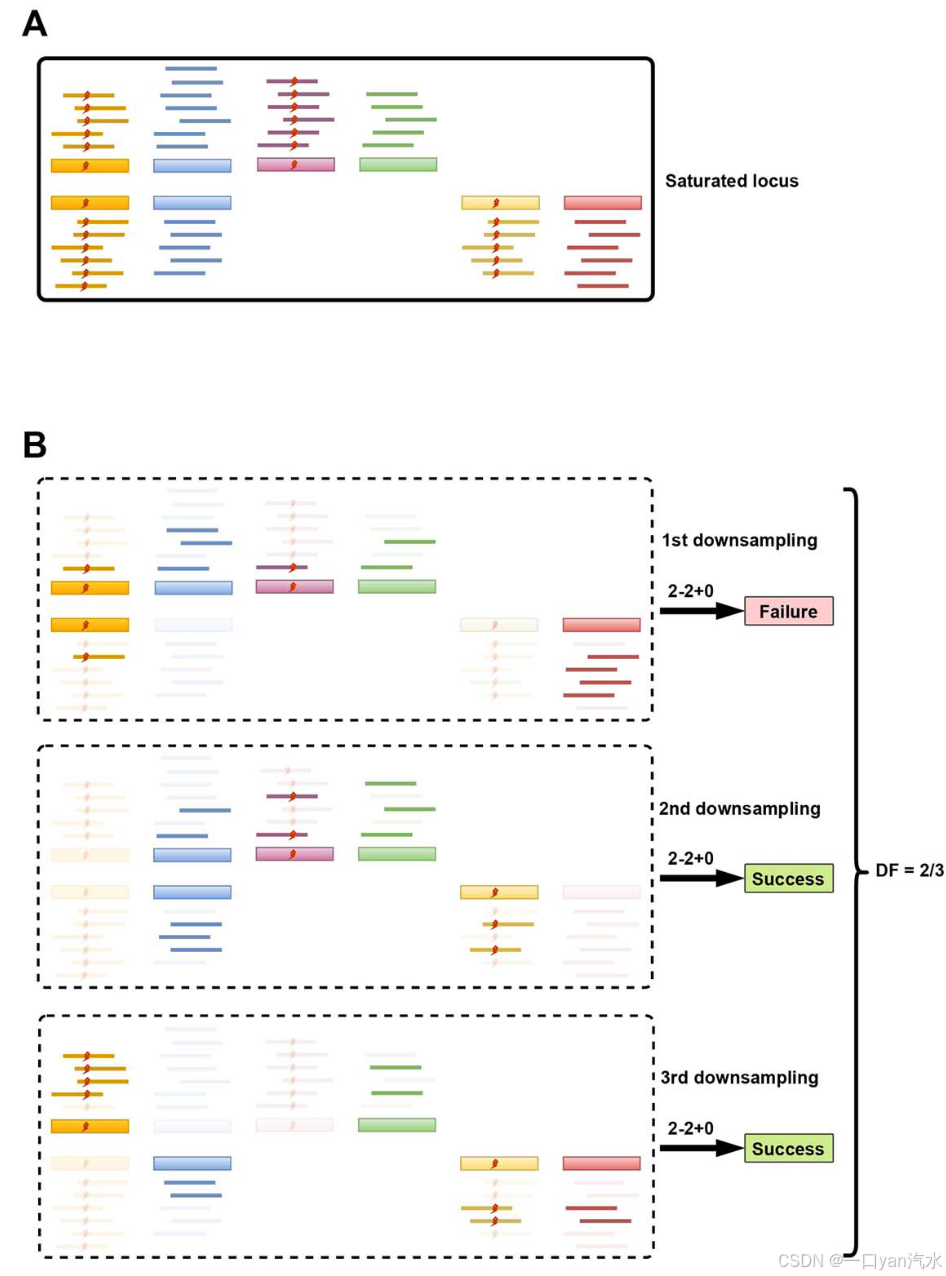
DELFMUT 的参数 DELFMUT 是为双工测序中的单基因座建模而设计的,其参数主要分为以下三类:(i) 饱和测序状态下,测序深度 DEPsatu、模板数量(重复深度)Tsatu、双链 DNA 模板比例 s、读数级链偏差 Rbias 和模板级链偏差 Tbias;(ii) 突变频率 VAF 和突变的突变检测规则 c - n1+n2;(iii) 下采样的位点目标深度 DEPtarget。这些参数的精确定义和数值如下:
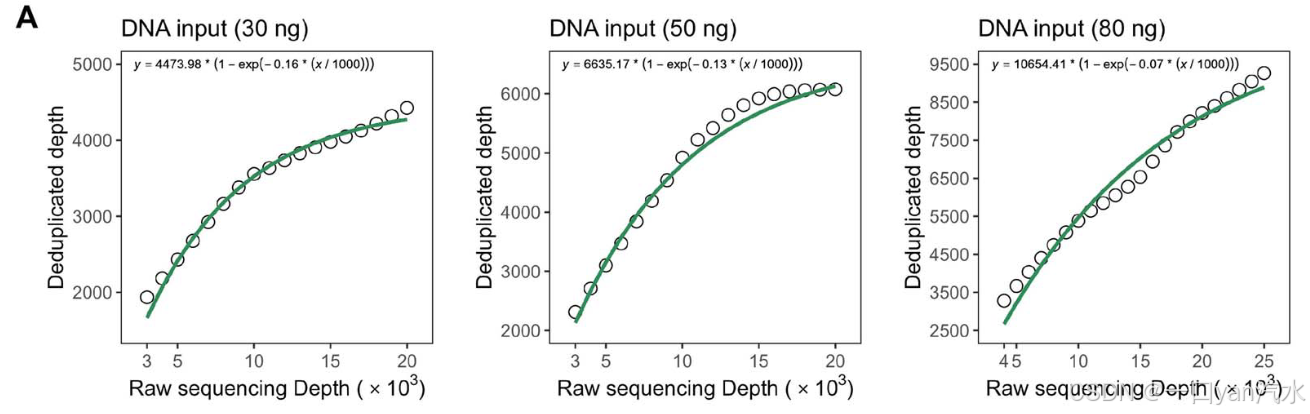
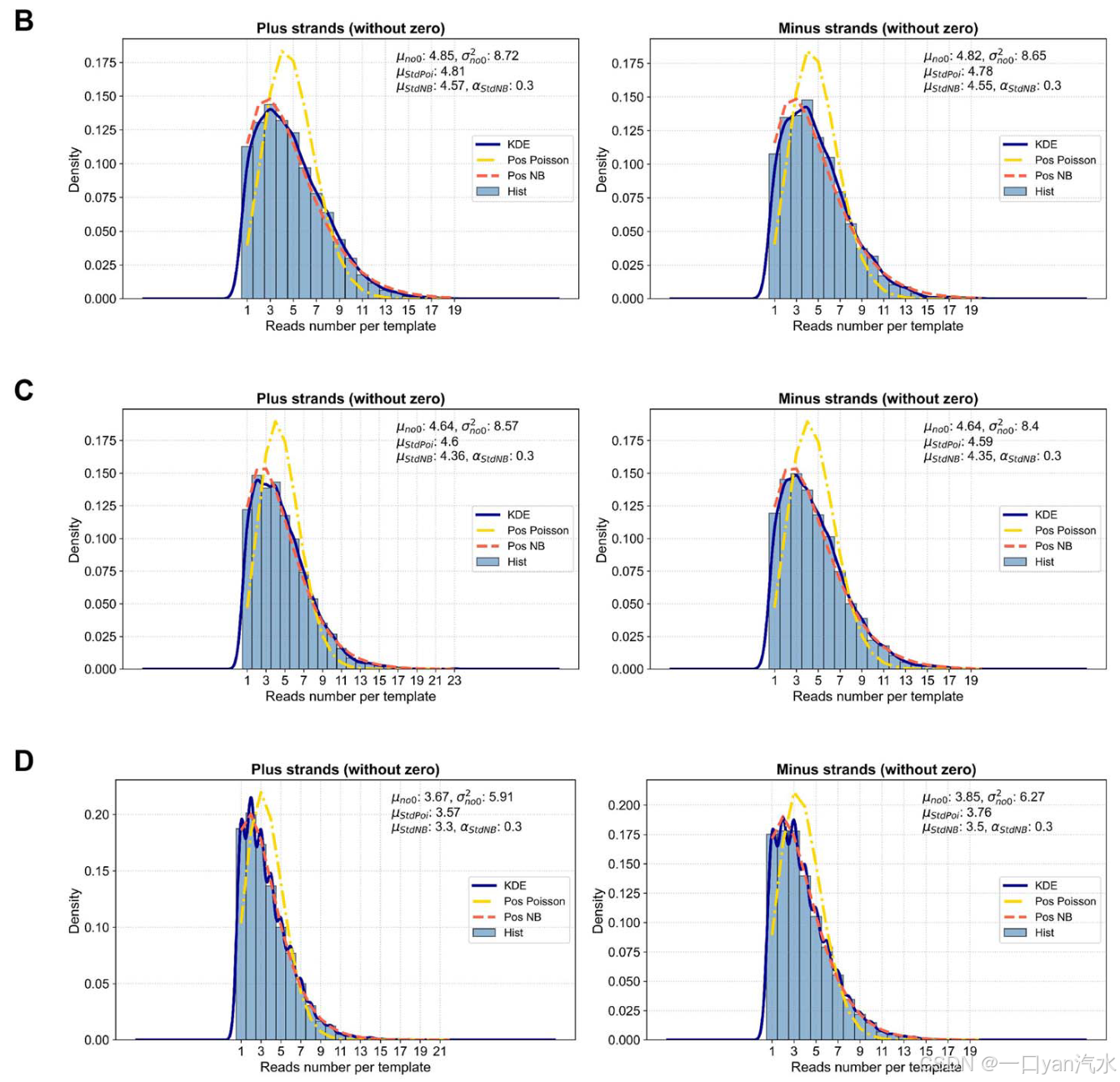
(i) 饱和测序状态下的测序深度 DEPsatu 旨在确保测序达到饱和状态,即可检测的模板数达到饱和值 Tsatu。因此,DEPsatu 的值没有上限,只有下限。为了减少计算量,建议在饱和后将 DEPsatu 设为一个较小的值,该值必须大于目标下采样深度 DEPtarget。由于样本的测序深度和重复深度分别是所有位点测序深度和重复深度的平均值,因此我们可以用样本级的饱和测序深度和重复深度来表示位点级的饱和测序深度和重复深度。饱和测序状态下等离子体样本的重复深度 Tsatu 由 DNA 输入 MASS 和测序技术的捕获效率决定。在此,我们使用另一种方法来计算 Tsatu:在给定 DNA 输入 MASS 的情况下,实际测序样本的重复深度 y 与测序深度 x 之间的关系可以用渐近指数函数 y = a - (1 - e-b- x 1000 ) [30]来拟合。a'和'b'分别代表渐近指数函数的渐近值和曲率参数。当 x 接近无穷大时,可以得到饱和测序状态下的重复深度 Tsatu = a。在我们收集的双工测序数据中,输入 30、50 和 80 纳克 DNA 时,计算出饱和测序状态下的重复深度 Tsatu 分别为 3892、5772 和 9268(图 2A,参见补充文件)。在此,我们选择了现有样本数量最多的 DNA 输入量,以便更准确地估算 DELFMUT 的输入参数。
我们还可以预先确定饱和状态下的其他参数:双链 DNA 模板比例 s、读数级链偏置 Rbias 和模板级链偏置 Tbias,因为它们在饱和测序状态下都保持稳定。
(ii) 当前位点的突变频率为 VAF,成功检测该突变的要求是检测出至少 c 个突变模板,每个模板至少有 n1 + n2 个突变支持读数,缩写为 c - n1+n2。这里,n1 和 n2 分别代表突变模板的正向链和反向链所需的最少突变支持读数,或按相反顺序排列。换句话说,突变检测规则既限制了突变模板的数量(≥ c),也限制了突变模板的正向链(≥ n1 或≥ n2)和反向链(≥ n2 或≥ n1)的突变支持读数的数量。此外,我们假设 n1 ≥ n2。当 n2 = 0 时,包含 SSCS;当 n2 > 0 时,只保留 DCS。通过设置不同的突变检测规则,可以控制检测到的突变数量和可靠性之间的平衡。
(iii) DEPtarget 是当前位点的目标下采样深度。我们反复将 DEPsatu 读数向下采样至 DEPtarget 读数,以模拟指定测序深度 DEPtarget 的情况。
DELFMUT 的主要过程
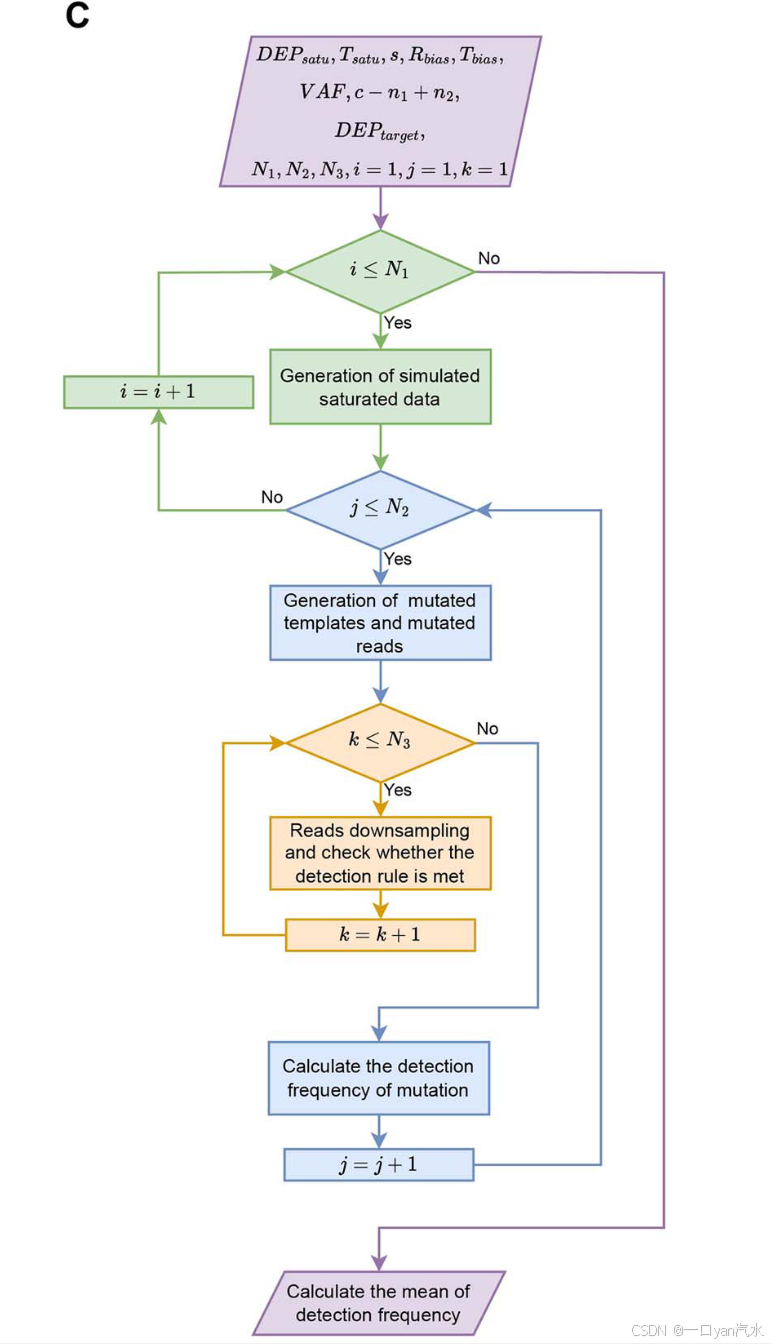
DELFMUT 主要包括四个步骤,步骤(i)-(iii)的重复次数分别记为 N1、N2 和 N3(图 1C)。主要步骤详述如下。
(i) 生成模拟饱和测序数据
(a) 初始化具有唯一 ID 的 Tsatu 模板,并按照 s : t : (1 - s - t) 的比例为其分配标签 "D"、"P "和 "N",分别代表双链模板、正向单链模板和反向单链模板。
(b) 我们使用零截断负二项分布来生成每个模板对应的读数数。将每个正向链模板对应的读数数的期望值 E1 和每个反向链模板对应的读数数的期望值 E2 作为零截断负二项分布的期望值 E,分别生成每个正向链模板和反向链模板对应的读数数。这里,我们采用了负二项分布的公式形式,其中使用了期望值 Estd 和超分散参数 α。通过求解截断为零的负二项分布 E 的期望值与标准负二项分布 Estd 的期望值之间的关系式,我们可以得出以下结果
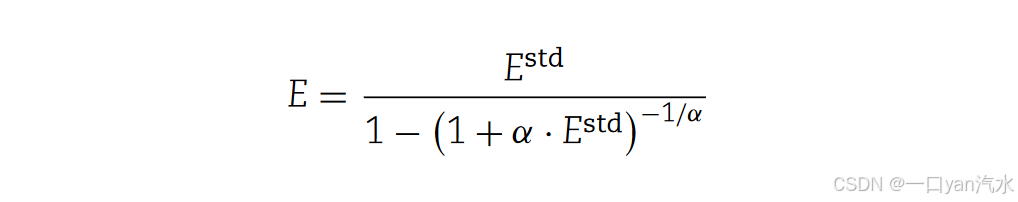
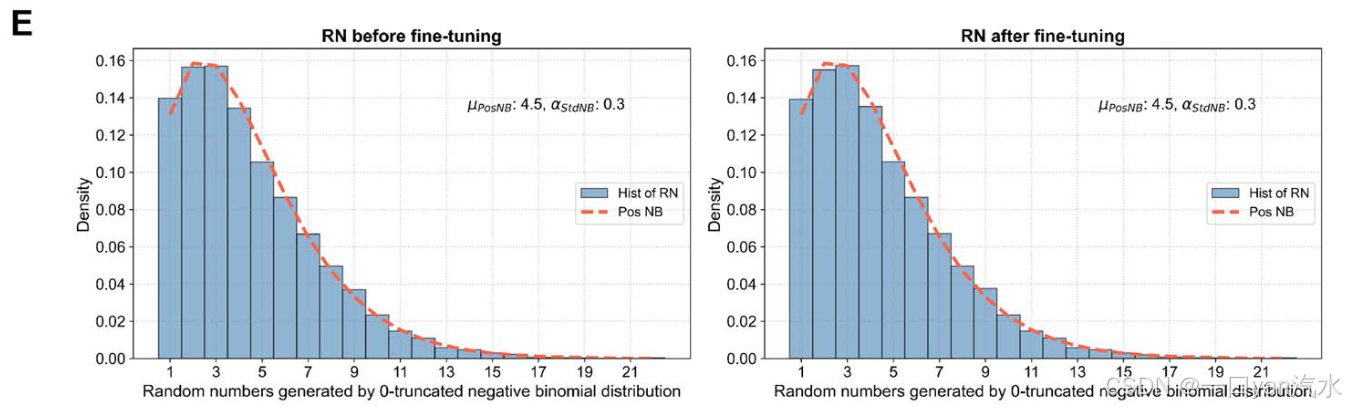
我们可以得到标准负二项分布的期望值 Estd 1 和 Estd 2(分别对应于 E1 和 E2)。标准负二项分布的参数α可通过预统计和参数调整预先得知,推荐值为 0.3(图 2B-D,参见补充文件)。我们为正向链模板生成 T1 随机数,为反向链模板生成 T2 随机数,分别遵循其特定的零截断负二项分布,并对其进行微调,使其和分别为 R1 和 R2(图 2E,参见补充文件)。这些随机数代表了每个模板对应的读数数量,根据这些数量关系,每个模板都会被分配一个支持该模板的读数 ID 列表。至此,我们已经生成了饱和测序状态下的模板和读数,并得到了它们之间的身份对应关系。
(ii) 突变模板和突变支持读数的生成 我们从 Tsatu 模板中随机选择 Tm satu = Tsatu - VA F 模板作为突变模板,并赋予它们突变标签。然而,计算得到的突变模板数量 Tm satu 通常不是整数,而是包含小数点后 v 位的正实数。我们可以通过控制⌊Tm satu ⌋和⌊Tm satu ⌋+1的取值频率来提高 DELFMUT 的精度和分辨率:⌊Tm satu ⌋ 出现 [(1 - v) - N2] 次,而 ⌊Tm satu ⌋ + 1 出现 [v - N2] 次。这里,v 代表 Tm satu 的小数位数,是四舍五入符号。详细推导过程见补充文件。
(iii) 对饱和测序数据的读数进行下采样,计算目标测序深度的突变检测频率 将读数下采样到给定深度 DEPtarget,重复 N3 次,然后利用检测规则 c - n1+n2 计算突变检测频率 fc-n1+n2。一般过程如下:
for k = 1toN3 do
(a) 从 DEPsatu 读数中随机抽取 DEPtarget 读数。
(b) 只保留 DEPtargets 采样读数。因此,模板、突变模板和突变支持读数的数量都将减少。
(c) 计算 Tm 突变模板中符合 n1+n2 规则的突变模板 x 的数量(即突变模板的每条正向链的突变支持读数满足≥ n1 或≥ n2,每条反向链的突变支持读数满足≥ n2 或≥ n1)。
end for
计算 N3 下采样中的突变检测频率 fc-n1+n2,检测规则为 c - n1+n2。
( iv )重复步骤( i ) - ( iii )以获得突变的平均检测频率重复上述步骤( i ) - ( iii )分别N1、N2和N3次,取步骤( iii )中获得的突变检测频率fc - n1 + n2的均值作为突变检测概率的估计值。步骤( i ) - ( iii )的重复是一个嵌套循环(图1C ):最内层的N3个循环是通过重复下采样来计算突变检测频率fc - n1 + n2;中间的N2循环是重复和随机地生成突变模板和支持突变的reads;外层N1环是整个过程的重复:从开始模拟饱和测序数据的产生,到突变模板和reads的产生,再到下采样。因此,这3个嵌套循环共进行N1 * N2 * N3的下采样。通过计算N1 * N2检测频率fc - n1 + n2的平均值fc - n1 + n2,可以估计在给定DNA输入MASS、测序深度DEPtarget和检测规则c - n1 + n2的情况下,突变频率为VAF的突变的检测概率。N1、N2和N3的值越高,DELFMUT的输出越准确。但考虑到DELFMUT的计算量和性能之间的平衡,当3个参数都取100时,可以得到相对稳定可靠的结果。因此,在本文的实验中,N1、N2和N3均设置为100。
DELFMUT支持同时输入饱和测序状态下的多个去重深度(与DNA输入量相对应)、多个突变频率VAF、多个检测规则和多个目标深度,可分别以数组的形式直接输入。DELFMUT可以输出这些输入参数的所有组合的突变检测频率。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









