




- 最新版本: 1.0.0
- 可以对着视频教程学习和使用:然而还没录呢, 关注B站等我更新

R包介绍
开发背景
- WGCNA是转录组或芯片表达谱数据常用得分析, 可用来鉴定跟分组或表型相关得模块基因和核心基因
- 但其步骤非常之多, 每次运行起来很是费劲, 但需要修改的参数并不多
- 所以完全可以编写成一站式流程, 直接从表达谱和表型得到所有结果
- 另外得到模块基因后, 后续还有PPI和Cytoscape的分析
功能简介
- 一站式实现WGCNA, 只需要输入表达谱和分组或表型文件, 即可完成分析
- 保存各种结果文件, 如每个模块基因、hub基因、显著性结果、MM/GS评分等等
- 可视化内容: 散点图、相关性热图重绘、相关性蝴蝶图
- 下游分析: Cytoscape的MCODE和cytoHubba结果的提取和可视化
安装
-
没有安装包的需要付费获取安装包:
- 直接添加微信转账100, 发送fastR(依赖包)和fastWGCNA的压缩包
- 如果购买过R包fastGEO, 可以半价购买fastWGCNA包!
- 很多资料后面都会慢慢涨价, 早买会便宜很多!
- 购买后可进入交流群,后续版本更新和问题答疑都在群里进行
-
R包fastGEO目前售价调整为100, 以前为20, 逐步涨价到50, 现在是100
-
两包合购限时优惠150, 限时多久看心情
-
所有R包随着功能增加价格必然会上调, 早买早享受, 且免费获取最新版本
# 依赖包
install_if_missing <- function(pkg, repos = getOption("repos")) {
if (!requireNamespace(pkg, quietly = TRUE)) {
install.packages(pkg, repos = repos)
}
}
install_if_missing("WGCNA")
install_if_missing("linkET")
install_if_missing("dplyr")
install_if_missing("pdftools")
install_if_missing("UpSetR")
install_if_missing("VennDiagram")
install_if_missing("gridExtra")
# 如果不存在fastR包或版本过低则进行安装
file_pkgs = grep("^fastR.*\\.gz$", dir(), value = TRUE)
if(!require("fastR", quietly = TRUE) || packageVersion("fastR") < strsplit(file_pkgs, "_|\\.tar")[[1]][2]){
install.packages(file_pkgs, repos = NULL, upgrade = FALSE, force = TRUE)
}
# 如果不存在fastWGCNA包或版本过低则进行安装
file_pkgs = grep("^fastWGCNA.*\\.gz$", dir(), value = TRUE)
if(!require("fastWGCNA", quietly = TRUE) || packageVersion("fastWGCNA") < strsplit(file_pkgs, "_|\\.tar")[[1]][2]){
install.packages(file_pkgs, repos = NULL, upgrade = FALSE, force = TRUE)
}
========================================
欢迎使用 fastWGCNA (版本 1.0.0)
- 作者: 生信摆渡
- 微信: bioinfobaidu1
- 帮助文档: help(package = fastWGCNA)
========================================
功能演示
library(fastWGCNA)
# 准备数据, 没有购买fastGEO的可以联系我购买或跳过此步
# 此步仅是数据准备, 也可以自行准备, 然后运行 run_WGCNA
library(fastGEO)
========================================
欢迎使用 fastGEO (版本 1.7.0)
- 作者: 生信摆渡
- 微信: bioinfobaidu1
- 帮助文档: help(package = fastGEO)
========================================
run_WGCNA
-
一站式运行WGCNA, 需要输入表达谱(expM)和分组向量(group)或表型数据框(pheno)
-
其他重要参数
- dir: 输出文件夹
- case_name: 实验组的名称
- pheno_name: 表型的名称, 会在图中显示
- ntop: 选取方差最大的前多少基因构建网络, 越少运行速度越快。当模块数或结果基因较少时, 可以调多一点, 默认5000
- npower: 软阈值计算的最大值, 默认是20, 一般就够了, 样本太少时可能需要调到30才会满足构建网络的需求
- cut: 去除离群样本的聚类数的高度, 大于该高度的会被去除, 默认不去
- soft: 选择的软阈值, 默认使用推荐的软阈值, 也可手动指定
- zlim: 相关性热图的相关性范围, 当相关性不高时, 范围调窄点会更好看些
- minModuleSize: 每个模块最小的基因数, 默认30, 最低了
- deepSplit: 数值越大(通常0-4),模块划分越精细,会产生更多更小的模块, 默认2
- reassignThreshold: 基因重新分配的阈值, 影响模块的纯度,较高的阈值可能保留更多模糊归属的基因, 默认0
- mergeCutHeight: 基于模块间的相关性进行合并,值越大,合并的模块越多, 默认0.25
- corr_cutoff: 判定模块显著性的相关系数, 默认为0, 只要P值<0.05就行, 最低阈值
- pvalue_cutoff: 判定模块显著性的P值, 默认0.05
- MM_cutoff: MM阈值, 默认0.8
- GS_cutoff: GS阈值, 默认0.3
-
如果没有得到显著的模块, 可以调整以下参数:
- ntop调大, 最大为nrow(expM), 即所有基因
- deepSplit调大
- mergeCutHeight调小
-
如果没有得到hub基因或太少, 可以调整以下参数:
- MM调低,最小为0.3
- GS调低,最小为0.3
obj = download_GEO("GSE30999", out_dir = "test/00_GEO_data_GSE30999")
INFO [2025-08-27 11:17:17] Step1: Download GEO data ...
INFO [2025-08-27 11:17:17] Querying dataset: GSE30999 ...
- Use local curl
- Found 1 GPL
- Found 170 samples, 39 metas.
- Writting sample_anno to test/00_GEO_data_GSE30999/GSE30999_sample_anno.csv
- Normalize between arrays ...
- Successed, file save to test/00_GEO_data_GSE30999/GSE30999_GPL570.RData.
INFO [2025-08-27 11:17:44] Step2: Annotate probe GPL570 ...
INFO [2025-08-27 11:17:45] Use built-in annotation file ...
- All porbes matched!
- All porbes annotated!
INFO [2025-08-27 11:17:46] Removing duplicated genes by method: max ...
INFO [2025-08-27 11:18:29] Done.
gruop_list = get_GEO_group(obj, group_name = "source_name_ch1", "NL" = "Control", "LS" = "Psoriasis")
expM = gruop_list$expM
group = gruop_list$group
hd(expM)
Type: data.frame Dim: 22881 × 170
GSM767976 GSM767978 GSM767980 GSM767982 GSM767984
A1BG 2.085353 2.079059 2.086824 2.081765 2.082029
A1BG-AS1 3.634412 2.953882 4.084853 3.958588 4.157088
A1CF 2.433706 2.395676 2.432235 2.381176 2.414382
A2M 13.489853 12.870176 12.957765 13.594529 13.878676
A2M-AS1 5.659882 4.673235 4.218735 5.400971 6.311412
table(group)
group
Control Psoriasis
85 85
# expM和group可自行准备
WGCNA_res = run_WGCNA(expM, group, case_name = "Psoriasis", pheno_name = "Psoriasis", dir = "test/GSE30999_WGCNA")
Select top 5000 genes with largest variation ...
Check data integrity ...
This a good data, nothing to do!
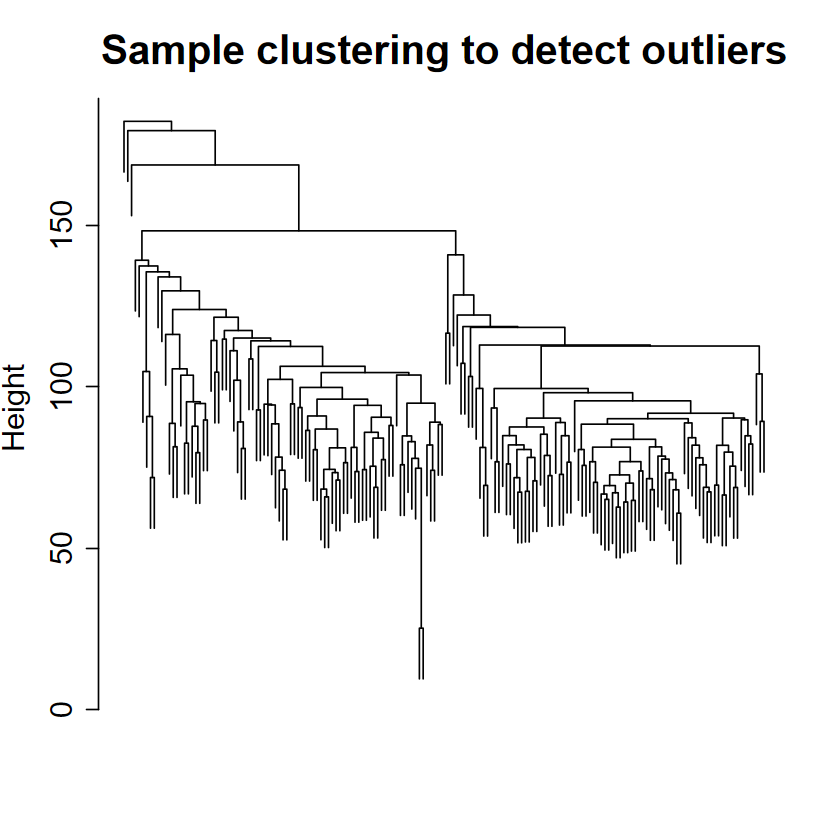
Detect outlier samples ...
Show_outlier samples
Remove 0 sample(s)
Cluster with pheno
Determine soft threshold ...
Warning message:
"executing %dopar% sequentially: no parallel backend registered"
Not specify soft threshold, using estimated: 6
Scale independence
Mean connectivity
Build modules ...
Plot Cluster dendrogram ...
Plot eigengene dendrogram ...
Show correlation and P-value between modules and phenos ...
Plot butterfly corrplot ...
Convert pdf to png ...
- 输出对象
str(WGCNA_res)
List of 12
$ datExpr : num [1:170, 1:5000] 2.27 8.36 2.68 2.62 9.24 ...
..- attr(*, "dimnames")=List of 2
.. ..$ : chr [1:170] "GSM767976" "GSM767978" "GSM767980" "GSM767982" ...
.. ..$ : chr [1:5000] "SERPINB4" "S100A12" "TCN1" "XIST" ...
$ datTraits :'data.frame': 170 obs. of 2 variables:
..$ Psoriasis: num [1:170] 0 0 0 0 0 0 0 0 0 0 ...
..$ Control : num [1:170] 1 1 1 1 1 1 1 1 1 1 ...
$ MEs :'data.frame': 170 obs. of 14 variables:
..$ MEblue : num [1:170] -0.0477 -0.169 -0.1571 -0.1265 -0.0532 ...
..$ MEgreen : 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









