







做常规分析关注 gene expression和phenotype
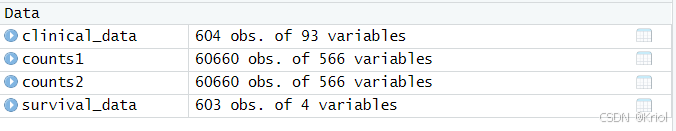
counts2有60660基因,566样本。
setwd("C:/Users/Fengsj/Desktop/R/analysis")
##读取数据----
library(data.table)
##读取counts数值
counts1 <- fread("./data/TCGA/TCGA-HNSC.counts.tsv.gz")
####bug处理
to_delete <- grep(pattern = "_PAR_Y", x = counts1$Ensembl_ID)
counts1 <- counts1[!to_delete,]
###bug处理
library(tidyverse)
##还原counts数值
counts1 <- column_to_rownames(counts1,"Ensembl_ID") ##把列转换成数据集的行名
counts2 <- 2^counts1 -1 ##把别人处理过的数据还原成counts
counts2 <- round(counts2) ##counts不能有小数,取整
survival_data <- fread("./data/TCGA/TCGA-HNSC.survival.tsv.gz") ##读取生存数据
clinical_data <- fread("./data/TCGA/TCGA-HNSC.clinical.tsv.gz") ##读取临床表征数据,不一定用
##数据预处理--过滤数据----
#过滤没有生存数据的样本
expr_sample <- colnames(counts2) ##获取表达数据的列名
survival_sample <- survival_data$sample ##获取生存数据的列名
#view(expr_sample)
#view(survival_sample)
#因为表达数据count2样本为566,生成数据survivaldata样本为603,所以要取交集
#只有数组可以取交集
valid_sample <- intersect(expr_sample,survival_sample)
#把这些样本的表达数据和生存数据提取出来
counts3 <- counts2[,valid_sample] #[行,列],表达数据提取
#生存数据的样本即不是列名也不是行名,而是内容,需要处理
surv_dat1 <- column_to_rownames(survival_data,"sample") ##将列变成行名
surv_dat1 <- surv_dat1[valid_sample,] #生存数据提取
#过滤非肿瘤样本
sample_type <- str_split(colnames(counts3), pattern = '-',n=4,simplify = TRUE) ##分割函数(分割位置,分割符,分割个数,分割后把所有数据组合在一起)
sample_type <- as.data.frame(sample_type) ##把对象转换成数组
sample_type <- sample_type[,"V4"] ##只取第四列,因为只有第四列才是代表不同的样本类型
unique(sample_type) ##看sampletype里有哪些类型,11A是正常组织,其他是肿瘤组织,要把11A都去除掉
sample_group <- cbind(sample_type) #cbind()按列结合
sample_group[,1] <- ifelse(sample_group[,1]=="11A","normal","tumor") #(若第一列等于11A赋值normal,否则tumor)
##经过之前counts3 <- counts2[,valid_sample]和urv_dat1 <- surv_dat1[valid_sample,]的处理,counts3样本排序和surv_data样本排序是一样的
##sample_group是按counts3操作的,他的排序也和他们一致
##直接把sample_group拼在他们后面是一一对应的
rows <- sample_group[,1]=="tumor" ##是肿瘤组织的行赋值给rows
counts4 <- counts3[,rows] ##把counts3即表达数据中的非肿瘤样本剔除
surv_dat2 <- surv_dat1[rows,] ##把生存数据中的非肿瘤样本剔除
##过滤完数据后,进行数据转化----
#获取所有基因的ID,并转换为基因名
gene_id <- rownames(counts4) ##表达数据的行名就是基因id
gene_id1 <- str_split(gene_id,pattern = '[.]',n=2,simplify = TRUE)[,1] ##基因id后面的小数点不要,用分割函数进行分割,分割后只要第一列
library(org.Hs.eg.db) ##将基因ID转化为基因名,这是人类基因数据库的一个包
#对应的geneid转化成genename
gene_name <- mapIds(org.Hs.eg.db, gene_id1,"SYMBOL","ENSEMBL") ##(数据库,需转化的数据集,SYMBOL是org.Hs.eg.db这个数据库里对于基因名的标志,匹配数据库里的数据源)
gene_name <- cbind(gene_name) ##把genename转化成数组(数据框)
gene_name <- as.data.frame(gene_name)
#view(gene_name)
#把genename放到表达数据里的行名里
counts5 <- counts4
counts5 <- as.data.frame(counts5)
rownames(counts5) <- gene_id1 ##把gene_id1赋给counts5的列名,确保后面代码counts5和genename的合并
#
#make.unique(gene_name[,1])
#rownames(counts5) <- gene_name[,1]
#any(duplicated(gene_name[,1]))
#
counts5 <- rownames_to_column(counts5, "gene_id") ##行名转化成列(要转化的文件,转化到的列的列名)
gene_name <- rownames_to_column(gene_name,"gene_id") ##行名转化成列
counts6 <- left_join(counts5,gene_name,by="gene_id") ##把counts5和genename合并,按照gene_id列合并
#genename到最后一列了,需要调整
counts6 <- relocate(counts6,gene_name,.after = gene_id)
counts6 <- column_to_rownames(counts6,"gene_id")
counts7 <- aggregate(.~gene_name, FUN=mean, data=counts6) ##不同基因id可能对应同一个基因,所以要计算均值进行合并
counts8 <- column_to_rownames(counts7,"gene_name") ##把基因名称放到列上


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









