






 本文介绍了生物信息学中的蛋白质同源检测和折叠识别概念,探讨了蛋白质在SCOP数据库中的分类,并概述了相关研究方法,包括基于比对、排序和判别式的方法。此外,讨论了数据集构建和评估标准,为初学者提供了入门知识。
本文介绍了生物信息学中的蛋白质同源检测和折叠识别概念,探讨了蛋白质在SCOP数据库中的分类,并概述了相关研究方法,包括基于比对、排序和判别式的方法。此外,讨论了数据集构建和评估标准,为初学者提供了入门知识。
前言
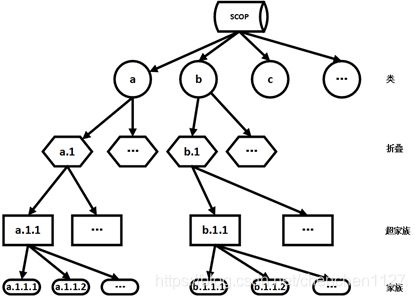
蛋白质同源性检测和折叠识别是近些年来生物信息学中的热点问题。通过蛋白质远同源性检测和折叠识别,能够初步推断未知蛋白质的结构和功能。其中,SCOP数据库[1]按照层级结构将蛋白质进行分类,从上到下依次是:类(class),折叠(fold),超家族(superfamily)和家(family)(如图1-1所示)。蛋白质远同源性检测的任务就是找出未知蛋白质所属的超家族。折叠识别的任务就是找出未知蛋白质所属的折叠类型。进一步根据所属的超家族和折叠类,推断出未知蛋白的结构和功能。由于具有远同源性的蛋白质之间序列相似度低(低于30%, 超家族层低于30%,折叠层低于20%),所以通过计算的方法来解决蛋白质远同源性检测问题和折叠识别一直是业内的一个难题。蛋白质远同源性检测研究当中中,相关方法主要能分为三类:基于比对的方法,基于排序的方法和基于判别式的方法[2]。蛋白质折叠识别的研究中,主要研究方法分为三类:基于比对的方法,基于机器学习的方法,基于集成的方法[3]。
目录
蛋白质同源检测
了解一个研究问题的三大步骤:what-->how-->why
- what?蛋白质同源检测的概念,蛋白质在SCOP数据库中分为近同源和远同源关系, 本文主要介绍蛋白质序列分析中远同源检测的相关入门知识。
- how? 如何去研究蛋白质同源检测的问题,数据集如何构建,当前的研究方法主要有哪些?
- why? 蛋白质同源检测的研究意义什么,本文不在赘述,相关知识请移步参考论文[2][3]。
蛋白质同源基本概念
同源(Homology):
Homology: the existence of shared ancestry between a pair of structures, or genes, in different species.
如果两个或多个结构具有相同的祖先,也就是它们由一个共同的祖先演化而来,则称它们同源(Homology)。
在生物信息中,同源主要是指序列上的同源,也就是用来说明两个或多个蛋白质或DNA序列具有相同的祖先。同源关系的强弱可以帮助了解物种间的亲缘关系。而且,同源的序列一般有相似的功能。序列中同源的部分也被称为保守的(conserved)。
蛋白质和DNA的同源性常常通过它们序列的相似性(Sequence similarity)来判定,相似性一般用检测序列和目标序列之间序列一致性(Percent identity)来表示。
相似性(Sequence similarity)是指序列比对过程中用来描述检测序列和目标序列之间相同DNA碱基或氨基酸残基顺序所占比例的高低。
近同源与远同源:
一般来说,当相似程度高于50%时,常推测检测序列和目标序列可能是近同源序列;
当相似性程度低于30%时,就难以确定其是否具有同源性,称其为远同源。
同源蛋白质:指进化上相关的蛋白质。即不同物种中具有相同或相似功能的蛋白质或具有明显序列同源性的蛋白质。
蛋白质远程同源性(remote homology)是指具有同源相似性性的蛋白质已经充分进化,不再具有较强的序列间的相似性。
蛋白质远程同源性检测(protein remote homology detect)的目的是预测蛋白质的家族信息,因为具有同源性的蛋白质通常具有相似的结构和功能,所以可以由已知蛋白质家族的结构和功能推测出新测定蛋白质的结构和功能
蛋白质家族(Protein family)和蛋白质超家族(Protein superfamilies):
蛋白质家族(Protein family)是指一组进化上相关的蛋白,具有同源性(来自于相同祖先),相似的结构及功能,显著的序列相似性。
蛋白质超家族(protein superfamily),一些蛋白质家族被归入更大的进化分支,基于结构机制的相似性,尽管其没有可以确定(显著)的序列同源性。简单而言,就是蛋白质超家族包括了更多进化相关的蛋白,虽然没说有同源性,但因为其结构或功能基本相似,也被归为一个大类。而蛋白质家族的同源关系是可以确定的,也就是关系上更加严格些。
注意: 蛋白质远同源检测就是在超家族层面的研究问题。
蛋白质同源检测方法简介
蛋白质远同源性检测研究当中中,相关方法主要能分为三类:基于比对的方法,基于排序的方法和基于判别式的方法[2].
基于比对的方法(Alignment method)
核心思想在于计算目标蛋白和折叠已知的模板蛋白之间的比对分数,将靶蛋白指定为具有最高比对评分的模板蛋白的折叠。侧重于检测局部和全局成对序列的相似性。为了提高灵敏度,在基于序列-序列比对方法的基础上进一步提出了基于谱的比对方法方法,包括序列-谱比对方法[4]和基于谱-谱比对方法,例如隐马尔可夫模型(HMM)[5]和马尔可夫随机场(MRF)。
基于排序方法(Ranking method)
核心思想在于把蛋白质远同源性检测看作是一个信息检索问题[4]。然后根据算法,把数据库中已知结构和功能的蛋白质与未知的查询蛋白质按照同源关系从近到远排序输出。其中排序算法的关键在于如何计算查询蛋白质序列和数据库中已知蛋白质序列的同源关系。
基于判别式方法(Discrimination method)
核心思想是将蛋白质远同源性检测看作是蛋白质超家族层面上的分类问题。对于预测任务,基于判别式的方法首先将蛋白质按照同源关系的远近划分正负样本训练集和测试集。然后用训练集中的蛋白质训练基于机器学习的分类模型。这个分类模型的性能将由测试集来评估,同时,在实际应用中,未知蛋白质所属的超家族也将由这个分类模型进行预测。
目前的判别式模型主要包括基于传统机器学习的方法(SVM 等)和基于深度学习技术的方法(LSTM, CNN-BLSTM)。
蛋白质折叠识别
什么是蛋白质折叠识别?与蛋白质远同源检测的区别在哪里?
蛋白质折叠识别简单来说就是在蛋白质折叠层面识别蛋白质。与蛋白质远同源检测的不同在于,远同源检测是在蛋白质超家族层面的研究,折叠识别的问题更加困难,一是在数据集上,要求序列相似度低于20%,远同源的要求序列相似度低于(30%);二在研究折叠识别问题时要抛开远同源即超家族层面的影响,所以要求目标蛋白与模板蛋白可以属于同一折叠类别,但不能属于同一超家族或家族。
蛋白质折叠识别相关概念
蛋白质结构域(protein domain)
是蛋白质中的一类结构单元,是构成蛋白质(三级)结构的基本单元。有些球形蛋白的一条肽链,或以共价键相连的两条或多条肽链在空间结构上可以区分为若干个球状的子结构,其中的每一个球状子结构就被称为一个结构域。
蛋白质结构域与蛋白质完成生理功能有着密切的关系,有时几个结构域共同完成一项生理功能,有时一个结构域就可以独立完成一项生理功能,但是一个结构不完整的蛋白质结构域是不可能产生生理功能的。因此蛋白质结构域是蛋白质生理功能的结构基础,但必须指出的是,虽然蛋白质结构域与蛋白质的功能关系密切,但是蛋白质结构域和功能域的概念并不相同。
模体(motif)
表示具有特定功能的或作为一个独立结构域一部分的相邻的二级结构的聚合体,它一般被称为功能模体(functional motif)或结构模体(structural motif),相当于超二级结构(super-secondary structure)。模体和结构域一起组成了蛋白质的三级结构。结构模体作为结构域的组分,介于蛋白质二级结构和三级结构之间。
蛋白质识别方法简介
蛋白质折叠识别的研究中,相关方法主要分为三类:基于比对的方法,基于机器学习的方法和集成学习方法。
基于比对方法
同远同源相似。
基于机器学习方法
核心思想通过使用经典机器学习技术或深度学习技术将蛋白质折叠识别视为折叠层面的分类任务,包括二分类和多分类任务。基于机器学习方法首先将蛋白质按照是否属于目标折叠划分正负样本训练集和测试集。然后用训练集中的蛋白质训练基于机器学习的分类模型。这个分类模型的性能将由测试集来评估,同时,在实际应用中,未知蛋白质所属的超家族也将由这个分类模型进行预测[26]。使用判别框架进行折叠识别,其关键在于特征提取方法和机器学习分类器,所以这些方法是基于各种判别特征和强大的分类器构建的,例如支持向量机(SVM)、随机森林(RF)和深度学习技术。
集成方法
核心思想采用共识策略来集成多种识别方法。
数据库和基准数据集
1.SCOP数据库
SCOP数据库[1]按照层级结构将蛋白质进行分类,从上到下依次是:类(class),折叠(fold),超家族(superfamily)和家(family)(如图1-1所示)。蛋白质的远同源和折叠识别分类问题基本都是基于SCOP数据库来做。目前SCOP数据库已经更新到SCOPe(version 2.07),详细信息及资源下载参考:http://scop.berkeley.edu/ .
2.蛋白质同源检测基准数据集
基于SCOP的蛋白质远同源检测基准数据集要求数据集必须满足任意两个蛋白质之间的序列相似度低于30%。且其任务主要是是在超家族层面对蛋白质进行分类识别。因此其构建过程主要包括去冗余操作、正负样本数据集划分和划分后不平衡数据集的处理。
首先进行去冗余,具体步骤:
- 去除SCOP数据库中相似度大于95%的蛋白质序列;
- SCOP数据库中少于10条蛋白质的家族将会被去除,若某个超家族中只含有一个家族,那么在这种情况下,该蛋白质超家族无法模拟蛋白质远同源性检测,因此,这个超家族也将会被去除。
其次,正负样本数据集的划分:
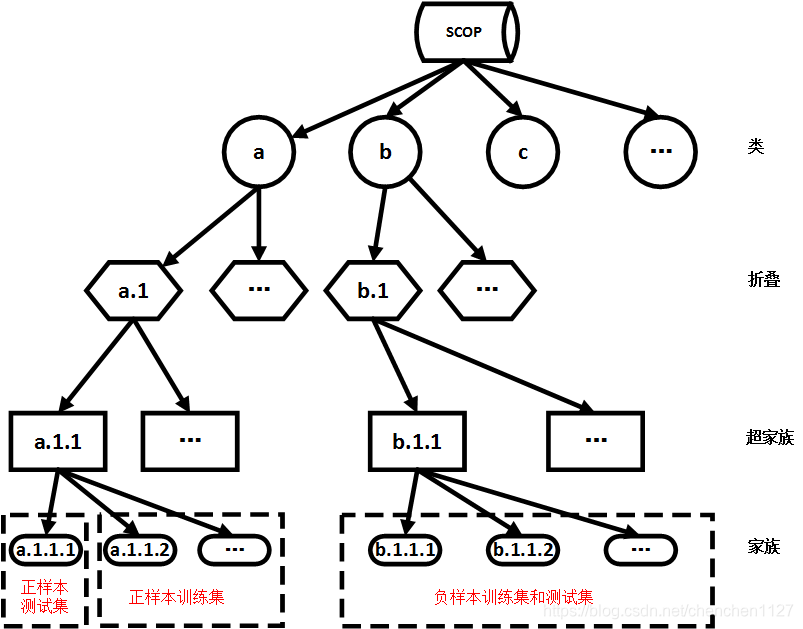
基础数据集中目标家族的训练集和测试集的定义由公式(2-1):
|
|
(2-1) |
其中,k表示第k个目标家族预测任务的数据集。![]() 表示第k个正样本测试集,它由第k个家族中的蛋白质组成。
表示第k个正样本测试集,它由第k个家族中的蛋白质组成。![]() 表示第k个正样本训练集,它由第k个家族所属的超家族中的所有蛋白质,除去第k个家族中的蛋白质所构成。从除了第k个超家族以外的每个超家族中,随机选取其中一个蛋白质家族,组合起来成为负样本测试集
表示第k个正样本训练集,它由第k个家族所属的超家族中的所有蛋白质,除去第k个家族中的蛋白质所构成。从除了第k个超家族以外的每个超家族中,随机选取其中一个蛋白质家族,组合起来成为负样本测试集![]() 。然后这些超家族中的其余家族的蛋白质作为负样本训练集
。然后这些超家族中的其余家族的蛋白质作为负样本训练集![]() 。构建方法示意图如2-1所示。
。构建方法示意图如2-1所示。
最后,不平衡数据集的处理:
基础数据集构建完成之后,由于训练集中的正样本和负样本极不平衡,常常呈百倍差异。而这种正负样本的数量差异会导致训练出的分类器在预测时,倾向于将未知样本预测成负样本,在现实应用中失去其预测意义。因此,Hochreiter采用了无监督的方法来拓展正样本训练集[48],拓展后的训练集的表示由公式(2-2)所示:
|
| (2-2) |
其中![]() 为第k个拓展的正样本训练集,其获取方式为:对于每一条属于
为第k个拓展的正样本训练集,其获取方式为:对于每一条属于![]() 的蛋白质,将其用PSI-BLAST工具在蛋白质数据库UniRef50数据库中搜索。搜索结果中E-value小于10的蛋白质序列作为拓展的正样本训练集。
的蛋白质,将其用PSI-BLAST工具在蛋白质数据库UniRef50数据库中搜索。搜索结果中E-value小于10的蛋白质序列作为拓展的正样本训练集。
3.蛋白质折叠识别基准数据集
此处等我自己的论文发表之后再补充。
图3-1 基准数据集构建方法示意图(此处等论文发表之后再上传)
References
[1]Murzin A G, Brenner S E, Hubbard T, et al. Scop: A Structural Classification of Proteins Database for the Investigation of Sequences and Structures[J]. J Mol Biol, 1995, 247(4): 536-540.
[2]李舒敏. 基于深度学习的蛋白质远同源性检测[D].哈尔滨工业大学,2018.
[3]Liu B, Li C, Yan K. DeepSVM-fold: Protein fold recognition by combining Support Vector Machines and pairwise sequence similarity scores generated by deep learning networks, Briefings in Bioinformatics;DOI: 10.1093/bib/bbz098.

















 2122
2122

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









