






之前我们有介绍过使用Banksy对多样本空间数据整合进行分析,有几个同学陆陆续续问了一些相关的问题,正好这些问题也是自己刚使用Banksy分析Xenium多样本数据中遇到过的一些疑惑。刚开始也看过许多公众号的介绍推文,但是都只是将github官方单个样本分析示例文档进行简单展示,没有关于多样本分析的详细介绍,以及Banksy中几个关键的函数具体是干什么的,几个关键参数啥意思,怎样根据目的设置参数等。这次将自己的项目实战经验和大家分享下,希望相互学习。
BANKSY 是一种对空间组学数据进行聚类的方法,它通过使用空间邻域特征的平均值和邻域特征梯度来增强每个单元的特征。通过整合邻域信息进行聚类,BANKSY 能够
-
改进噪点数据中的单元类型分配
-
区分按微环境分层的细微不同的细胞类型
- 识别共享相同微环境的空间域
Banksy的核心算法设计
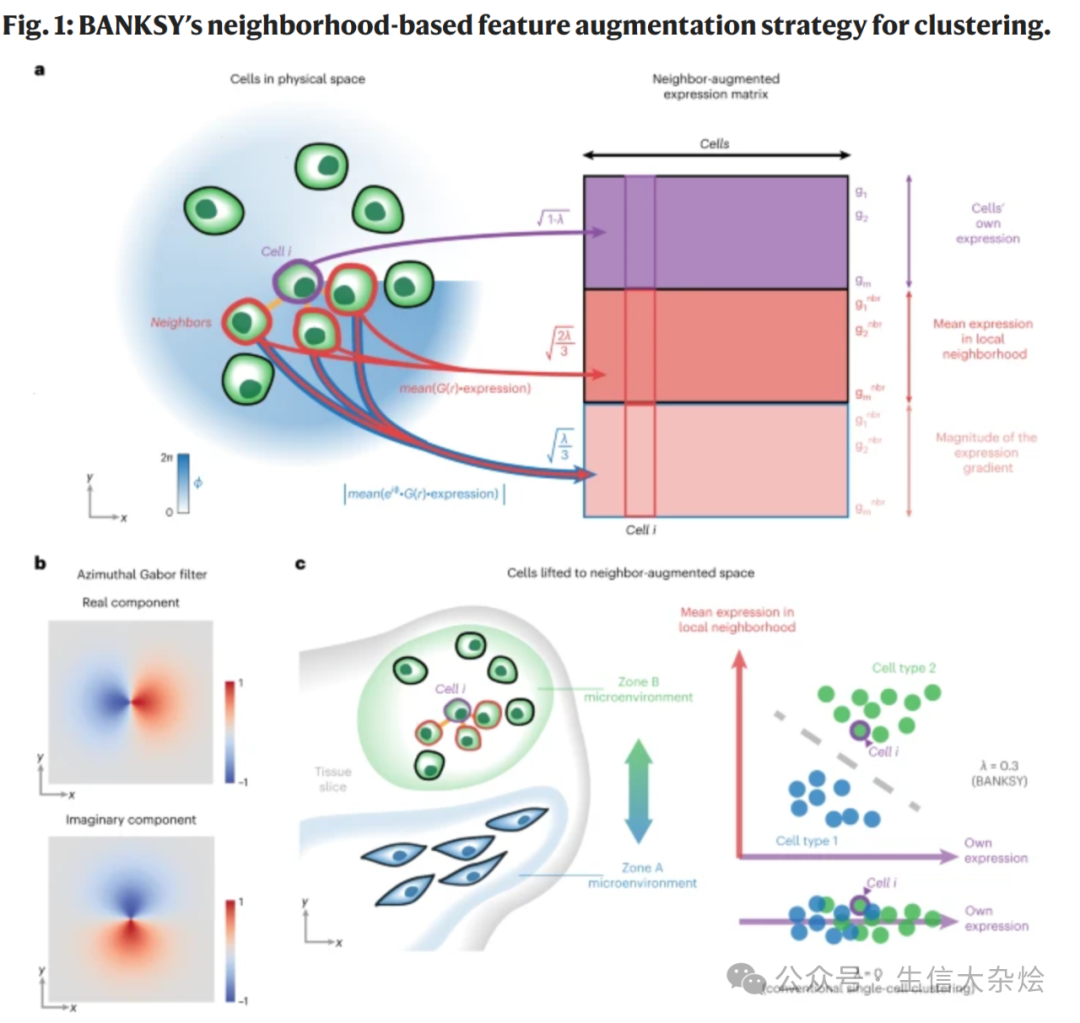
通过将每个细胞的特征与其空间近邻特征的平均值以及邻域特征梯度相结合来增强细胞的特征表示。它使用两个空间核来编码微环境的转录组纹理,一个基于每个细胞邻域中基因表达的加权平均值,另一个使用方位加布滤波器(AGF)。通过一个混合参数λ(取值范围为[0,1])来控制细胞自身转录组和邻域表达矩阵的相对权重。当λ较小时,强调细胞自身的转录组,使细胞根据细胞类型聚类;当λ增加时,强调邻域特征,使细胞根据组织域聚类。
一句话总结:Banksy在进行细胞聚类的时候不光使用单个细胞的转录本特征,还能够结合细胞邻域特征梯度来增强细胞的特征进行聚类,通过混合参数λ控制细胞邻域特征权重,λ=0表示只使用单细胞特征进行聚类,λ=1表示不考虑单细胞特征,只使用空间特征进行聚类。
有了上述背景知识,我们就可以解答之前的一些疑问了
1. 多个样本合并数据使用Banksy进行分析,为什么首先需要将样本的空间坐标进行错开?
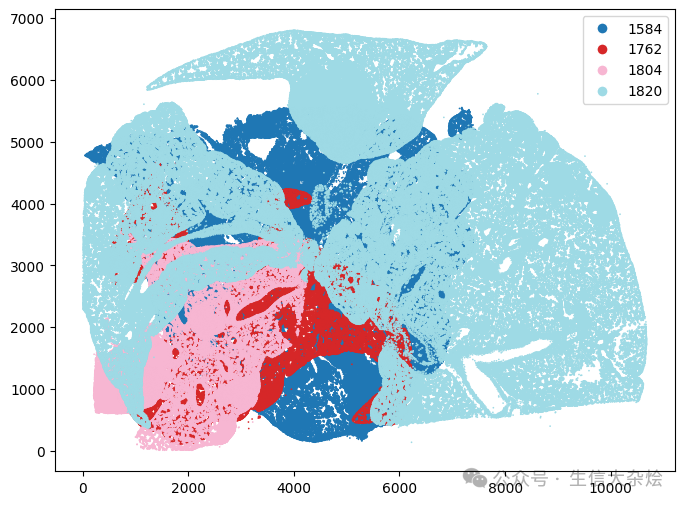
这里我们用4个小鼠肺腺癌样本Xenium数据作为示例,展示不经任何处理,这几个样本的坐标重叠情况,可以看到这些样本的坐标大部分都是重叠的,有前面的原理我们知道,Banksy需要根据每个样本中细胞空间坐标计算他们的最近邻细胞,构建空间邻域图,显然每个样本的细胞空间坐标是不能重叠的。
import scanpyimport pandas as psimport matplotlib.pyplot as pltfrom matplotlib.lines import Line2Dadata = sc.read_h5ad('../xenium_mouse_lung_adenocarcinoma.h5ad')#细胞数太多,这里随机抽20%数据进行演示sc.pp.sample(adata, 0.2)def plot_spatial_coordinates_samples(adata):adata.obs['x_pixel'] = adata.obsm['spatial'][:, 0]adata.obs['y_pixel'] = adata.obsm['spatial'][:, 1]# 为每个样本生成颜色unique_samples = adata.obs['sample'].unique()colors = plt.cm.tab20(np.linspace(0, 1, len(unique_samples)))color_dict = {sample: colors[i] for i, sample in enumerate(unique_samples)}sample_colors = [color_dict[s] for s in adata.obs['sample']]fig, ax = plt.subplots(figsize=(8, 6), dpi=100)ax.scatter(x=adata.obs['x_pixel'],y=adata.obs['y_pixel'],c=sample_colors,alpha=1,s=1.5,edgecolor='none')legend_elements = [Line2D([0], [0], marker='o', color='w', label=sample, markerfacecolor=color_dict[sample], markersize=8) for sample in unique_samples]ax.legend(handles=legend_elements)plot_spatial_coordinates_samples(adata)
2. 多个样本的空间坐标是怎么错开的
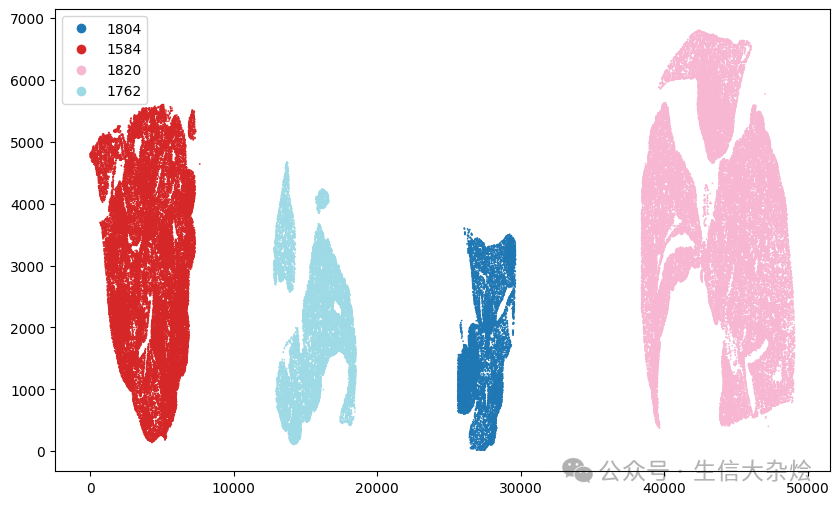
简单理解就是将多个样本沿x轴一次排开,中间间隔一定距离
adata.obs['x_pixel'] = adata.obsm['spatial'][:, 0]adata.obs['y_pixel'] = adata.obsm['spatial'][:, 1]# Staggeringcoords_df = pd.DataFrame(adata.obs[['x_pixel', 'y_pixel', 'sample']])coords_df['x_pixel'] = coords_df.groupby('sample')['x_pixel'].transform(lambda x: x - x.min())global_max_x = max(coords_df['x_pixel']) * 1.2#Turn samples into factorscoords_df['sample_no'] = pd.Categorical(coords_df['sample']).codes#Update x coordinatescoords_df['x_pixel'] = coords_df['x_pixel'] + coords_df['sample_no'] * global_max_x#Update staggered coords to AnnData objectadata.obs['x_pixel'] = coords_df['x_pixel']adata.obs['y_pixel'] = coords_df['y_pixel']
多个样本空间坐标错开后展示如下:
3. initialize_banksy函数的作用
initialize_banksy() 是 BANKSY 空间转录组分析工具中的关键初始化函数,用于为空间聚类准备数据结构。
核心作用:
-
构建空间邻域图
-
基于细胞的空间坐标计算每个细胞的邻居
-
使用
k_geom参数指定每个细胞考虑的最近邻居数量
-
-
计算空间权重
-
通过
nbr_weight_decay('scaled_gaussian' or 'reciprocal')控制邻居权重随距离衰减的速度
-
-
生成多尺度特征矩阵
-
创建从
m=0(仅自身基因表达)到max_m(包含多阶邻居)的特征矩阵
-
参数解析:
|
参数 |
作用 |
|---|---|
adata |
AnnData 空间转录组数据集 |
coord_keys |
空间坐标在 |
k_geom |
每个细胞考虑的最近邻居数量 |
nbr_weight_decay |
邻居权重衰减系数 |
max_m |
空间聚合的最大阶数 |
plt_edge_hist |
是否绘制细胞间距离分布直方图 |
plt_nbr_weights |
是否绘制权重随距离变化的曲线 |
4. generate_banksy_matrix函数的作用
generate_banksy_matrix() 是 BANKSY 工作流中的核心特征生成函数,其作用是根据不同的空间权重参数 λ 创建多尺度的空间增强特征矩阵。
核心作用:
-
生成多尺度空间特征
为每个 λ 值创建从m=0(原始表达)到max_m(多阶邻居聚合)的特征矩阵 -
空间信息融合
将基因表达与空间邻域信息按不同权重 (λ) 融合 -
构建聚类输入矩阵
生成最终用于聚类的特征矩阵
参数解析:
|
参数 |
作用 |
|---|---|
adata |
AnnData 对象 |
banksy_dict |
来自 |
lambda_list |
空间权重系数列表 |
max_m |
空间聚合的最大阶数 |
输出解析:
-
banksy_dict(更新后) -
banksy_matrix(核心输出)-
形状:
(n_cells, n_genes × (max_m+1) × n_lambda)
-
λ 参数的作用:
-
λ = 0:仅使用原始基因表达
-
λ = 0.2:弱空间权重
-
λ = 0.8:强空间权重
-
λ = 1.0:完全依赖邻居特征
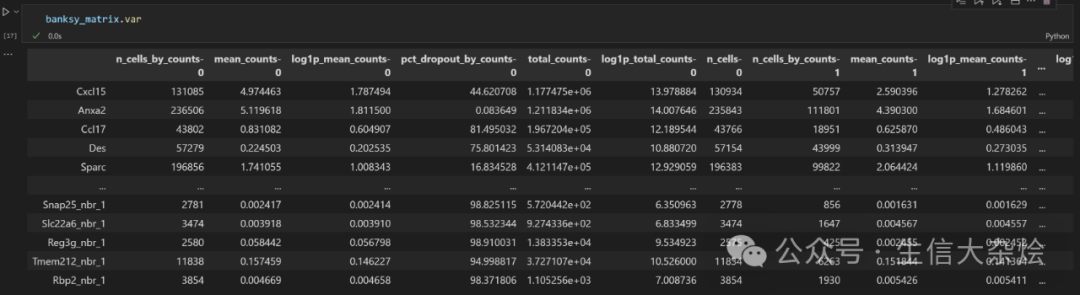
5. 我明明只用了前2000的HVG进行分析,为什么generate_banksy_matrix函数返回的banksy_matrix中有6000个基因?
因为前面我们说了,有个参数m,这个空间最大阶数我们设置的值为1,他会从m=0开始,到m=1各计算一遍基因表达,我们输入的原始2000个基因,加上m=0算的2000个,再加上m=1算的2000个,所以就是6000个,相当于每个基因有三个,只是基因名标注不一样了,如下
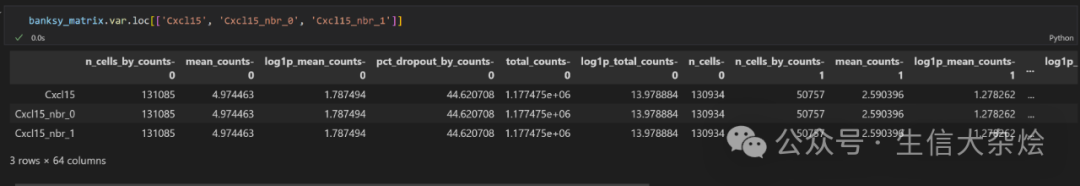
不过这些都不重要,我们最终只是要得到聚类的meta data,原始的基因表达不会变的。只要将Banksy得到的X_umap和聚类信息加到我们原始的adata中即可。
6. 怎样将Banksy得到的X_umap和聚类信息加到我们原始的adata中
我们根据实际使用,将多样本数据使用Banksy封装成相应函数,首先使用Banksy构建空间邻域、计算空间权重、生成多尺度空间特征、PCA降维、harmony批次矫正、生成最终用于聚类的特征矩阵使用leiden进行降维聚类,得到Banksy后的banksy_adata,只要将banksy_adata的meta信息复制给原始adata即可。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









