数据准备
rm(list = ls())
#打破下载时间的限制,改前60秒,改后10w秒https://www.baidu.com/?tn=88093251_70_hao_pg
options(timeout = 100000)
options(scipen = 20)#不要以科学计数法表示
#数据准备
corhorts = c("GSE6364", "GSE7305", "GSE51981", "GSE120103")
library(tinyarray)
f = "exp_groups.Rdata"
if(!file.exists(f)){
dats = lapply(1:4, function(i){geo_download(corhorts[[i]]) })#数据下载
par(mfrow = c(2,2))#图片以2*2的形式存在,也可以不写
for (i in 1:4){boxplot(dats[[i]]$exp[,1:10])}#检查数据是否有异常
dats[[1]]$exp = log2(dats[[1]]$exp+1)#对未取log的数据取log
dats[[2]]$exp = log2(dats[[2]]$exp+1)
探针注释
#探针注释
find_anno("GPL570",install = T)#产生探针注释的代码语言
#前三个数据均为GPL570平台,最后一个为GPL6480
library(hgu133plus2.db)
ids <- toTable(hgu133plus2SYMBOL)
get_gpl_txt("GPL6480",destdir = getwd(), download = T)
#可能因为网络报错,下载较慢,可从得到的连接自行下载并放入工作目录
soft = rio::import("GPL6480.txt")
#rio::import()从文件中读入 data.frame。Rdata、RDS 和 JSON 输入文件格式除外,返回最初保存的对象,而不更改其类型。
#检查筛选探针注释,去除空白行
colnames(soft)
id4 = soft[,c("ID","GENE_SYMBOL")]
k = id4$GENE_SYMBOL==""
table(k)
id4 = id4[!k,]
colnames(id4) = c("probe_id","symbol")
dats[[4]]$exp[1:4,1:4]
分组信息
### 分组信息
#View(dats[[4]]$pd)
library(stringr)
k = str_detect(dats[[1]]$pd$description,"normal")
table(k)
g1 = ifelse(k,"normal","patient")
g1 = factor(g1,levels = c("normal","patient"))
k = str_detect(dats[[2]]$pd$title,"Normal")
table(k)
g2 = ifelse(k,"normal","patient")
g2 = factor(g2,levels = c("normal","patient"))
table(dats[[3]]$pd$`tissue:ch1`)
keep =dats[[3]]$pd$`tissue:ch1` %in% c("Early Secretory Endometrial tissue",
"Proliferative Endometrial tissue",
"Mid-Secretory Endometrial tissue")
dats[[3]]$pd = dats[[3]]$pd[keep,]
dats[[3]]$exp = dats[[3]]$exp[,keep]
g3 = str_remove(dats[[3]]$pd$`tissue:ch1`," Endometrial tissue")
g3 = factor(g3,levels = c("Early Secretory",
"Mid-Secretory",
"Proliferative"))
table(g3)
keep = dats[[4]]$pd$`sample group:ch1` %in% c("Endometrium from Stage IV Ovarian Endometriosis of fertile women",
"Endometrium from Stage IV Ovarian Endometriosis of Infertile women")
table(keep)
dats[[4]]$pd = dats[[4]]$pd[keep,]
dats[[4]]$exp = dats[[4]]$exp[,keep]
g4 = ifelse(str_detect(dats[[4]]$pd$`sample group:ch1`,
"Infertile"),"Infertile","fertile")
g4 = factor(g4,levels = c("Infertile","fertile"))
table(g4)
groups = list(g1 = g1,
g2 = g2,
g3 = g3,
g4 = g4)
表达矩阵
### 表达矩阵,将exp行名由探针名转换为基因名
exps = list()
for(i in 1:3){
exps[[i]] = trans_array(dats[[i]]$exp,ids)#为微阵列或rnaseq表达矩阵转换行名
}
exps[[4]] = trans_array(dats[[4]]$exp,id4)
for(i in 1:4){
print(identical(ncol(exps[[i]]),length(groups[[i]])))
}#测试两个对象是否完全相等;此处为exp的样本(列数)数与groups的样本数(长度)是否相等?
save(exps,groups,file = "exp_groups.Rdata")
}
load(f)
转录因子的筛选
#转录因子的筛选
##文章下载了(1)来自推断序列结合偏好目录(CISBP)数据库(Weirauch等人,2014)的1639个tf
#(2)来自人类转录因子数据库(Human TFDB)的1665个tf (Hu等人,2019)
#(3)来自人类转录因子数据库(Lambert等人,2018)的1639个tf
#取三个数据库中tf的交集
a = rio::import("Homo_sapiens_TF.txt")$Symbol
b = readLines("TF_names_v_1.01.txt")
d = riRo::import("TF_Information.txt")$TF_Name
d = unique(d)#d内由重复的,需去重复
library(tinyarray)
TFs = intersect_all(a,b,d)#取交集
save(TFs,file = "TFs.Rdata")
TFv = list(a,b,d)
names(TFv) = c("AnimalTFDB","The Human Transcription Factor","CIS-BP")
draw_venn(TFv,"")#来自{tinyarray}
#用其他R包画Venn图
##library(gplots);venn(TFv)
差异分析
#差异分析
load("exp_groups.Rdata")
ids = lapply(1:2, function(i){
data.frame(probe_id = rownames(exps[[i]]),
symbol = rownames(exps[[i]]))
})
dcp = get_deg_all(exps[[1]],groups[[1]],ids[[1]],logFC_cutoff = 0)
dcp2 = get_deg_all(exps[[2]],groups[[2]],ids[[2]],logFC_cutoff = 0)
dcp[[3]]
dcp2[[3]]
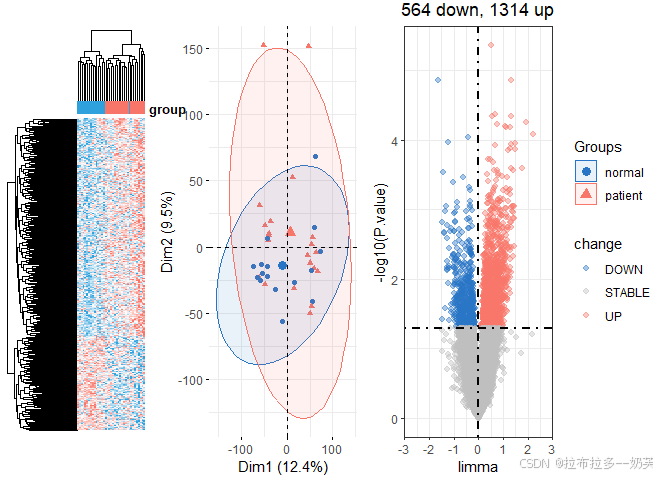
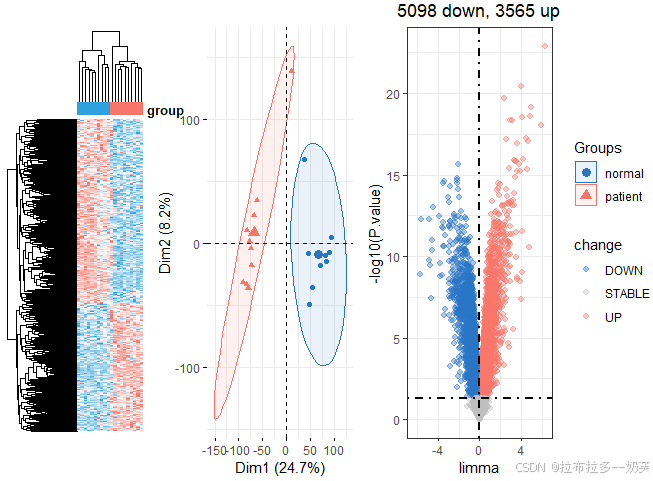
差异基因
#差异基因
corhorts#脚本01内命名过的文件数据集
#得出上、下调基因及其Venn图
up = list(dcp[["cgs"]][["deg"]][["up"]]$upgenes,
dcp2[["cgs"]][["deg"]][["up"]][["upgenes"]],
TFs)
names(up) = c(corhorts[1:2],"TFs")
draw_venn(up,"up")
down = list(dcp[["cgs"]][["deg"]][["down"]]$downgenes,
dcp2[["cgs"]][["deg"]][["down"]]$downgenes,
TFs)
names(down) = c(corhorts[1:2],"TFs")
draw_venn(down,"down")
up_gene = intersect_all(up)
down_gene = intersect_all(down)
up_gene
down_gene
draw_heatmap(exps[[1]][c(up_gene,down_gene),],groups[[1]],cluster_cols = F,show_rownames = T)
n = exps[[1]][c(up_gene,down_gene),]
g = groups[[1]]
n = n[,order(g)]#order(g)——所有数据按照g升序排列
g = sort(g)#对向量或因子(部分)进行升序或降序排序。
draw_heatmap(n,g,cluster_cols = F,show_rownames = T)
draw_heatmap(exps[[2]][c(up_gene,down_gene),],groups[[2]],show_rownames = T)
#上述基因为自己代码运算得到
#为接下来方便文献复现,将原文中分析得到的差异基因带入分析当中
##原文中的差异基因
library(stringr)
up_gene2 = str_split("FOXO4, MLXIPL, SPI1, ZNF205, VDR, POU2F2, ZNF793, ETV7, ZNF418, ZNF671, ARX, MSC, FOXC1, ZNF646, TBX1, ATF5, THAP6, POU6F1, MAFG, FOXK1, MYRF, THAP3, ZNF182, THAP2, ATF4, MEOX1, LYL1, NR5A1, RUNX2, GLI3, DMRTA1, ESR2, ZNF524, NR0B1, NFATC2, BATF, THRA",", ")[[1]]
down_gene2 = str_split("SOX5, IRF6, EN2, HOXB8, ZBTB18, SREBF1, ZNF764, OVOL2, SOX13, FOXF1, PLAGL2, ZNF750, MYSM1, ZSCAN16, ZIC3, ZNF513, ZNF236",", ")[[1]]
up_gene2
down_gene2
##画热图
n = exps[[1]][c(up_gene2,down_gene2),]
g = groups[[1]]
n = n[,order(g)]
g = sort(g)
draw_heatmap(n,g,cluster_cols = F,show_rownames = T)
draw_heatmap(exps[[2]][c(up_gene2,down_gene2),],groups[[2]],show_rownames = T)
table(c(up_gene,down_gene)%in% c(up_gene2,down_gene2))
##对比自己分析和文章分析的差别
k = rownames(exps[[1]])%in% c(up_gene2,down_gene2)
table(k)
table(c(up_gene2,down_gene2) %in% ids[[1]]$symbol)
k1 = rownames(exps[[3]]) %in% up_gene2;table(k1)
k2 = rownames(exps[[3]]) %in% down_gene2;table(k2)
k3 = rownames(exps[[4]]) %in% up_gene2;table(k3)
k4 = rownames(exps[[4]]) %in% down_gene2;table(k4)
draw_boxplot(exps[[3]][k1,],groups[[3]],drop = T)
draw_boxplot(exps[[3]][k2,],groups[[3]],drop = T)
draw_boxplot(exps[[4]][k3,],groups[[4]],drop = T)
draw_boxplot(exps[[4]][k4,],groups[[4]],drop = T)
##富集分析
ER=quick_enrich(c(up_gene2,down_gene2),kkgo_file = "g2.Rdata")
names(ER)
library(patchwork)
G$go.dot
G$kk.dot
write.table(c(up_gene2,down_gene2),file = "deg.txt",quote = F,row.names = F,col.names = F)
#做ROC曲线
install.packages("pROC")
library(pROC)
library(ggplot2)
#BATF在GSE6364
m1 <- roc(groups[[1]], exps[[1]]["BATF",],plot=T)
g1 <- ggroc(m1,legacy.axes = T)
g1 + theme_minimal() +
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")
auc(m1)
#RUNX2在GSE6364
m2 <- roc(groups[[1]], exps[[1]]["RUNX2",],plot=T)
g2 <- ggroc(m2,legacy.axes = T)
g2 + theme_minimal() +
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")
auc(m2)
#BATF在GSE7305
m3 <- roc(groups[[2]], exps[[2]]["BATF",],plot=T)
g3 <- ggroc(m3,legacy.axes = T)
g3 + theme_minimal() +
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")
auc(m3)
#RUNX2在GSE7305
m4 <- roc(groups[[2]], exps[[2]]["RUNX2",],plot=T)
g4 <- ggroc(m4,legacy.axes = T)
g4 + theme_minimal() +
geom_segment(aes(x = 0, xend = 1, y = 0, yend = 1),
colour = "grey", linetype = "dashed")
auc(m4)



























 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









