







 本文是msprime的学习笔记,介绍了msprime的主要功能,包括模拟树、重组、突变、历史样本等。msprime是一个高效、简便的溯祖树模拟工具,用于模拟不同进化场景。文中详细讲解了模拟树的实现,突变和重组的影响,以及如何处理历史样本。此外,还探讨了msprime在群体结构和群体统计学中的应用,包括模拟群体结构和使用重组图谱。
本文是msprime的学习笔记,介绍了msprime的主要功能,包括模拟树、重组、突变、历史样本等。msprime是一个高效、简便的溯祖树模拟工具,用于模拟不同进化场景。文中详细讲解了模拟树的实现,突变和重组的影响,以及如何处理历史样本。此外,还探讨了msprime在群体结构和群体统计学中的应用,包括模拟群体结构和使用重组图谱。
msprime
msprime是经典的ms模拟软件的重实现,关于msprime的安装和全部使用帮助如下:
msprime简介
msprime的主要功能是在一系列进化场景下高效、方便地为样本生成溯祖树(coalescent trees)。该库是Hudson开创性的ms程序的一个重新实现,旨在最终重现其所有功能。msprime在一些重要方面与ms不同:
1.更高效,相对ms模拟软件以及其他基于与重组模型近似结合的模拟软件来讲,其在内存使用和模拟时间上都有着绝对优势,尤其是当样本量很多时。
2.简便,msprime主要用于通过Python API简化运行和分析模拟。首先编写一个脚本来生成想要运行的命令行参数,然后使用fork shell进程来运行模拟,解析结果,就获得所需的谱系。
3.sprime不使用ms中的Newick trees进行交换,因为它们在存储空间和生成和解析它们所需的时间方面非常低效。相反使用tskit库,它允许我们有效地存储和处理非常大规模的模拟结果。
msprime安装
关于msprime的环境搭建及安装过程都在以下链接中,在此我们不过多赘述。
msprime安装指导
msprime教程
模拟树
import msprime
tree_sequence = msprime.simulate(sample_size = 6,Ne = 1000)
tree = tree_sequence.first()
print(tree.draw(format = "unicode"))

注意,由于数据为随机模拟,在重现模拟时几乎不可能与本笔记结果相同。
首先,利用简单直观的代码,我们模拟了有效群体大小为1000的二倍体的六个群体的系谱树,并描绘出来。在这里,msprime的模拟结果由tskit包表示,而simulate()函数会返回到一个tskit.TreeSequence对象,当模拟涉及重组时,通过该对象可与相关性树高效联合。在此案例中,由于没有设置重组率recombination_rate,其默认为0,所以用first()函数访问序列中唯一一棵树,然后用tskit.Tree.draw()函数绘制出对该序列树的简单描述。
- Genealogical trees record the lines of descent along which genomes have been inherited. Since diploids have two copies of each autosomal chromosome, diploid individuals contain two such lines of descent: the simulation above provides the genealogical history of only three diploids.关于此处为什么只能表示三个二倍体,没有考虑明白,在后续过程中继续学习。
tskit对树的表示方式与寻常不同,在大多数库中,节点表示为内存中的一个对象,而节点间的联系表示对象间的指针。然而在tskit中,所有节点都是数字符integers。
u = 2
while u != tskit.NULL:
print("node {}: time = {}".format(u, tree.time(u)))
u = tree.parent(u)
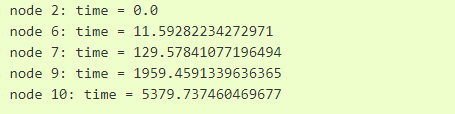
在此代码中,我们用u指示模拟出的树的节点,绘制从该节点到根节点的过程(根节点的数值是 -1 ,但此处使用符号常量更具可读性)。使用time()函数获取每个节点的时间,对应模拟过程中结合发生的时间。我们还可以使用branch_length()方法获得节点与其父节点分支的长度:
print(tree.branch_length(6))

节点6的分支长度约为118代,为节点7的129代减去节点6的11代。同样可以用total_branch_length()获取分支总长度:
print(tree.total_branch_length)

重组
为了对重组影响下的基因组序列进行建模,msprime API 有两个参数来控制执行simulate()函数。length 参数指定模拟序列的长度,并且是一个浮点数,因此可以在序列的任何位置发生重组和突变,其默认值为1.0。recombination_rate 参数指定每个单位长度上的染色体互换率,默认值为0。关于这两个参数的详细用法,可以在API文档中查阅:
API 文档
接下来,我们模拟一个10kb区域内树,其中每代每个碱基的重组率为2×10−8,二倍体有效种群大小为1000:
tree_sequence = msprime.simulate(
sample_size = 6, Ne = 1000, length = 1e4, recombination_rate = 2e-8)
for tree in tree_sequence.trees():
print("_" * 20)
print("tree {0}: interval = {1}".format(tree.index, tree.interval))
print(tree.draw(format="unicode"))
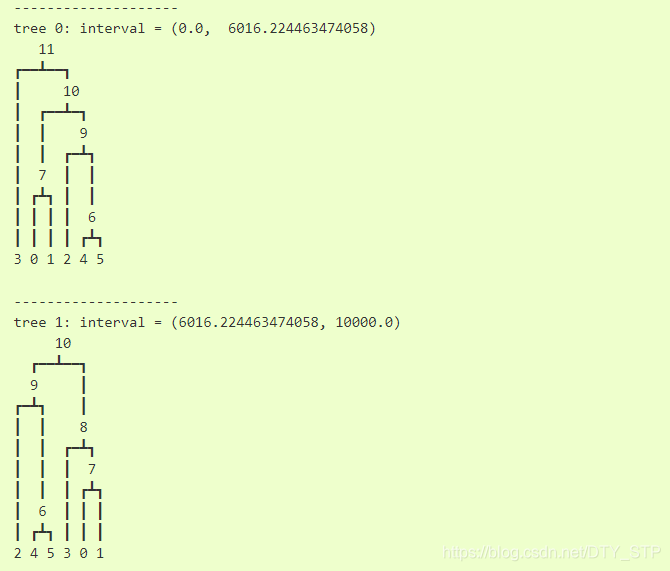
在这个例子中,我们利用tskit.TreeSequence.trees()的方法迭代整个10KB序列中所产生的树结构。然后利用tskit.Tree.index和 tskit.Tree.interval属性,按顺序描绘出每棵树的覆盖区域。例如,此例中两棵树分别占据了整个序列的约前6kb和后4kb。但不同树中的相似结构(节点的数字符相同)是不同的。
注意:
不要将
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









