




脊髓性肌萎缩症(Spinal Muscular Atrophy,SMA)是一种常见的常染色体隐性神经肌肉疾病,由生存运动神经元1基因(SMN1)突变引起。SMN1基因突变中约40%为无义或移码突变,这类突变会引入提前终止密码子(Premature Termination Codon,PTC)。无义介导的mRNA衰变(Nonsense-Mediated mRNA Decay,NMD)是一种高度保守的真核质量控制途径,能够降解含有PTC的mRNA,防止潜在有毒蛋白的积累。然而,SMN1基因中不同PTC突变触发NMD的能力存在差异,需要进一步评估。本文将基于minigene系统,对研究文献“Premature termination codons in SMN1 leading to spinal muscular atrophy trigger nonsense-mediated mRNA decay”进行深入解读。
研究者选取了三种导致SMA的SMN1无义突变:c.43C>T、c.683T>A和c.844C>T,分别位于外显子1、5和7。构建包含这三个突变的SMN1-minigene,并将其转染到HeLa细胞中。通过体内剪接分析、环己酰胺(CHX,NMD抑制剂)处理和siRNA介导的UPF1(NMD关键因子)敲低等方法,研究这些突变对NMD途径的影响。
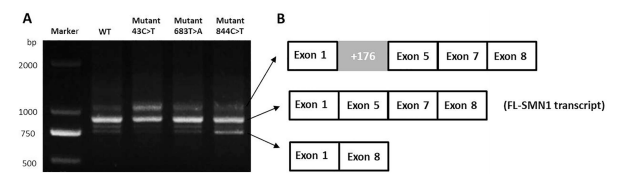
图1 minigene实验验证SMN1剪接变体的转录分析结果
野生型minigene(WT)仅产生正确剪接的转录本(FL-SMN1),即所有外显子(1、5、7和8)都被包含,内含子完全被移除。这表明构建的SMN1-minigene系统正常工作,能够产生正常的转录本。c.43C>T突变型minigene产生了两种不同的转录本,主要转录本是FL-SMN1。
次要转录本较长,经过测序分析发现包含了176bp的片段,该片段包含内含子1的155bp、XbaI限制位点TCTAGA以及内含子4的15bp。c.844C>T突变型minigene:产生了FL-SMN1转录本和一种较少见的较短转录本。经过测序分析,较短的转录本被鉴定为跳过了外显子5和7的转录本。
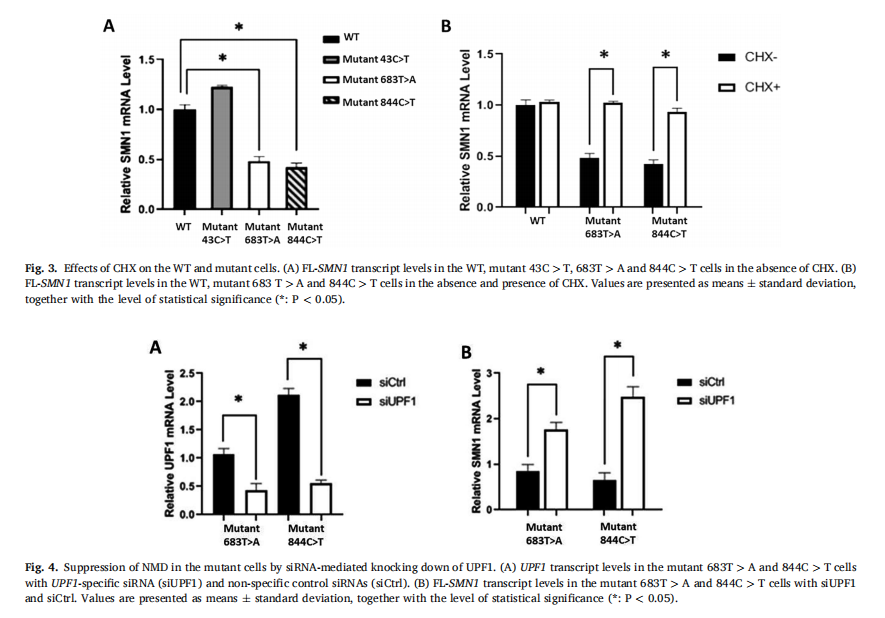
图2 NMD机制验证实验
经CHX(NMD抑制剂)处理后,突变细胞c.683T>A和c.844C>T中FL-SMN1的水平显著增加,说明NMD在这些细胞中活跃,且CHX能够抑制NMD,增加NMD底物转录本的水平。敲低UPF1(NMD关键因子)的表达可显著增加突变细胞c.683T>A和c.844C>T中FL-SMN1的转录本水平,进一步证实了这两个突变激活了NMD。而c.43C>T突变未触发NMD,其转录本水平不受CHX和UPF1敲低的影响。
本研究揭示了SMN1基因中不同PTC突变对NMD途径的影响,表明c.683T>A和c.844C>T突变可触发NMD,而c.43C>T突变则不能。这一发现对于理解SMA的发病机制具有重要意义。NMD在SMA的发病过程中可能扮演双重角色,一方面可能消除编码截短蛋白的转录本,防止其产生潜在毒性;另一方面,若截短蛋白保留部分正常功能,NMD的激活可能导致更严重的疾病表型。研究结果提示,在探索SMN1基因突变的致病机制时,应充分考虑NMD的影响。
参考文献
Zhang, Mengya, et al."Premature termination codons in SMN1 leading to spinal muscular atrophy trigger nonsense-mediated mRNA decay."Clinica Chimica Acta 530 (2022): 45-49.

















 1269
1269

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









