






孟德尔随机化,即使用遗传变异作为工具变量(IV),估计暴露对结果的因果效应。大多数 IV 方法都假设暴露与结果预期值之间的函数关系(暴露-结果关系)是线性的。但是一方面,线性假设可能并不成立;另一方面,人们也关心暴露-结果关系的形状,所以就要用到非线性孟德尔随机化。为了实现非线性孟德尔随机化,本篇文章介绍nlmr包的使用。
该包使用两种 IV 方法来研究暴露-结果关系的形状:分数多项式法和分段线性法。具体方法为,利用暴露的分布将人群划分为不同的层,并估算每个层的因果效应,即局部平均因果效应(LACE)。分数多项式法对这些 LACE 估计值进行meta回归;分段线性法估算一个连续的分段线性函数,其梯度就是每个分层的LACE估计值。 这两种方法都能很好地估计真实的暴露-结果关系,特别是当这种关系是分数多项式(分数多项式法)或片断线性(片断线性法)时。
首先向大家介绍包的作者在官网(GitHub - jrs95/nlmr: Non-linear Mendelian randomization)上分享的例程,该例程没有使用具体的个体层面数据,而是使用随机数示范了两个主要函数的用法。
install.packages("devtools")
library(devtools)
install_github("jrs95/nlmr")
library(nlmr)
#step1 生成模拟数据
epsx <- rexp(10000)#生成一个长度为10000的指数分布随机数向量,表示暴露除去IV预测部分的残差(se值)
u <- runif(10000, 0, 1)# 生成一个长度为10000的均匀分布随机数向量,表示混杂因素的影响。
g <- rbinom(10000, 2, 0.3)#生成一个长度为10000的二项分布(p=0.3)随机数向量,模拟工具变量。
epsy <- rnorm(10000)#生成一个长度为10000的正态分布随机数向量,表示结局除去IV预测部分的残差(se值)
ag <- 0.25 #工具变量g对暴露x的影响系数(β值)
x <- 1 + ag * g + u + epsx# 生成暴露x
y <- 0.15 * x^2 + 0.8 * u + epsy#生成结局y
#协变量
c1 = rnorm(10000)
c2 = rnorm(10000)
c3 = rbinom(10000, 2, 0.33)
c = data.frame(c1 = c1, c2 = c2, c3 = as.factor(c3))
### step2 nlMR分析
#fracpoly_mr 使用分数多项式进行 IV 分析
fp = fracpoly_mr(y, x, g, c, family = "gaussian", q = 10, d = "both", ci = "model_se", fig = TRUE)
summary(fp)
#piecewise_mr: 用分段线性模型进行IV分析
plm = piecewise_mr(y, x, g, c, family = "gaussian", q = 10, nboot = 50, fig = TRUE)
summary(plm)例程最终得到fp(分数多项式分析)和plm(分段线性模型分析)的图像如下:
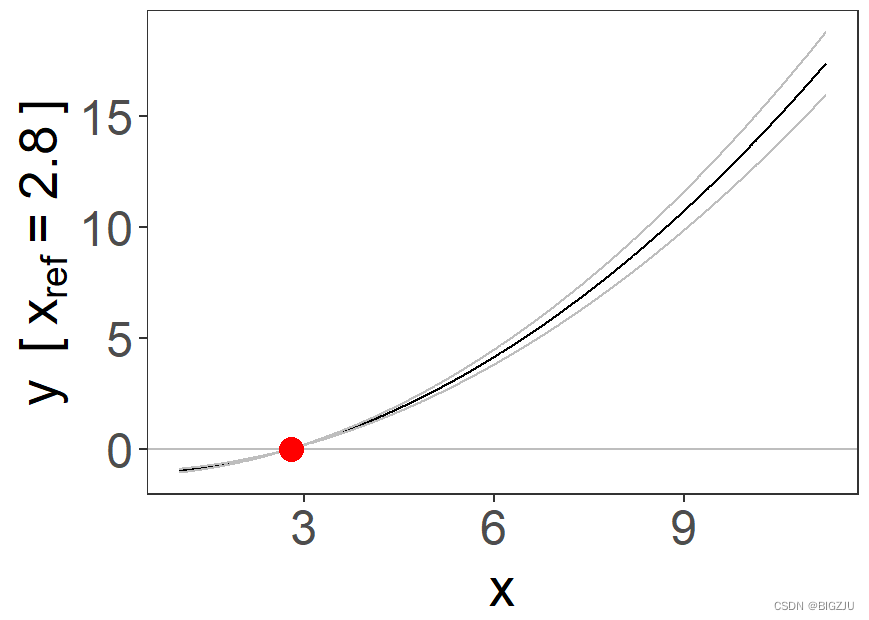
|
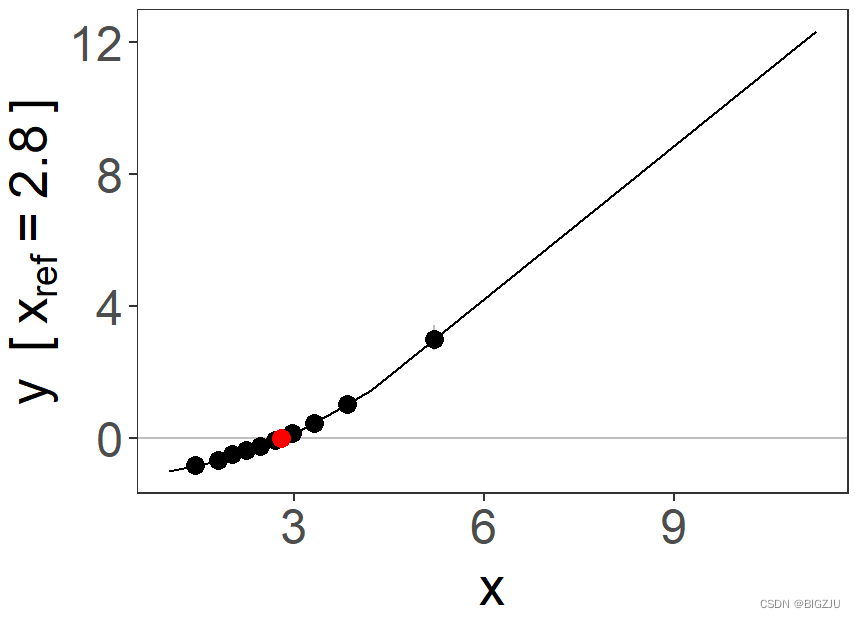
|
为了让大家对nlmr包的应用有更具体的理解,下面我以探究BMI与冠心病之间的因果关系为例,展示SNPs,β值,se值的获取。暴露和结局要用到个体水平的数据(比如ukb),此处仍以函数生成的形式展现,大家使用代码时改为个体水平的暴露和结局数据即可。
install.packages("devtools")
library(devtools)
install_github("jrs95/nlmr")
library(nlmr)
#比如,想探究BMI(id:ieu-a-2)与冠心病(id:ieu-a-7)之间的因果关系
#id可直接从数据库网站搜索(https://gwas.mrcieu.ac.uk/) 也可使用以下代码搜索
ao <- available_outcomes()
## 使用grep1抓取目标表型
ao[grepl("heart disease", ao$trait), ]
#查看完整结果后我们发现冠心病GWAS id为 ieu-a-7
###第一步 从GWAS数据库中获得SNPs、暴露/结局与遗传变异的β值(SNP对暴露/结局的效应量),se值(β值的标准误),此处展示利用TwoSampleMR包的方法
remotes::install_github("MRCIEU/TwoSampleMR")
library(TwoSampleMR)
## 定位数据位置,将路径赋予bmi2_file
bmi2_file <- system.file("extdata/bmi.csv", package="TwoSampleMR")#extdata/bmi.csv是TwoSampleMR的自带数据
bmi1_data <- read.csv(bmi2_file)
##读取暴露数据
bmi_exp_dat <- read_exposure_data(
filename = bmi2_file,
sep = ",",
snp_col = "rsid",#SNP(单核苷酸多态性)标识符
beta_col = "effect",#效应大小(遗传变异对表型的影响程度)
se_col = "SE",#效应大小标准误差(Standard Error)
effect_allele_col = "a1",#效应等位基因
other_allele_col = "a2",#其他等位基因
eaf_col = "a1_freq",#效应等位基因频率(Effect Allele Frequency)
pval_col = "p-value",#p值(用于检验效应是否显著的统计量)
units_col = "Units",#效应大小单位
gene_col = "Gene",#基因名称
samplesize_col = "n"#样本量
)
##对暴露数据进行聚类处理
bmi_exp_dat <- clump_data(bmi_exp_dat,clump_r2=0.01 ,pop = "EUR")
#在遗传关联研究中,由于连锁不平衡(linkage disequilibrium)的存在,多个SNP可能会同时与某个表型关联。为了避免这种多重关联性,通常会对数据进行聚类处理,将高度相关的SNP聚为一类,只保留每个类中的一个代表性SNP。
# clump_r2=0.01:指定聚类时使用的r²阈值。如果两个SNP之间的r²(决定系数)大于这个值,它们将被视为高度相关,并只保留其中一个。
# pop = "EUR":指定人口群体为欧洲人群(European)。这是因为在不同的人群中,连锁不平衡的模式可能会有所不同,因此聚类处理时可能需要考虑人口群体的差异。
#结果:在30个变异中,有3个变异由于与其他变异存在连锁不平衡(LD)或未出现在连锁不平衡参考面板中而被移除
##提取结局GWAS数据
chd_out_dat <- extract_outcome_data(
snps = bmi_exp_dat$SNP,
outcomes = 'ieu-a-7'
)
##对数据进行预处理,使其效应等位与效应量保持统一
dat <- harmonise_data(
exposure_dat = bmi_exp_dat,
outcome_dat = chd_out_dat
)
##从dat数据集中获取nlmr包进行IV分析需要的参数
g <- as.numeric(factor(dat$effect_allele.exposure, levels = c("A", "T", "C", "G"), labels = c(0, 1, 2, 3))) #IV
ag <- dat$beta.exposure #工具变量对暴露的β值
epsx <- dat$se.exposure #暴露的se值
epsy <- dat$se.outcome #结果的se值
###第二步 需要从ukb中获取个体水平的暴露与结局数据,考虑到数据的可即性,这里利用简单函数生成的数据进行示例。
x <- 2 + ag * g + epsx# 生成暴露x,其中包括工具变量的影响。
y <- 0.15*x^2 + epsy#生成结果y,其中包括暴露x的非线性影响。
### Analyses
#fracpoly_mr 使用分数多项式进行 IV 分析
fp = fracpoly_mr(y, x, g, family = "gaussian", q = 5, d = "both", ci = "model_se", fig = TRUE)
summary(fp)
#piecewise_mr: 用分段线性模型进行IV 分析
plm = piecewise_mr(y, x, g, family = "gaussian", q = 5, nboot = 50, fig = TRUE)
summary(plm)
最终得到fp和plm的图像如下:
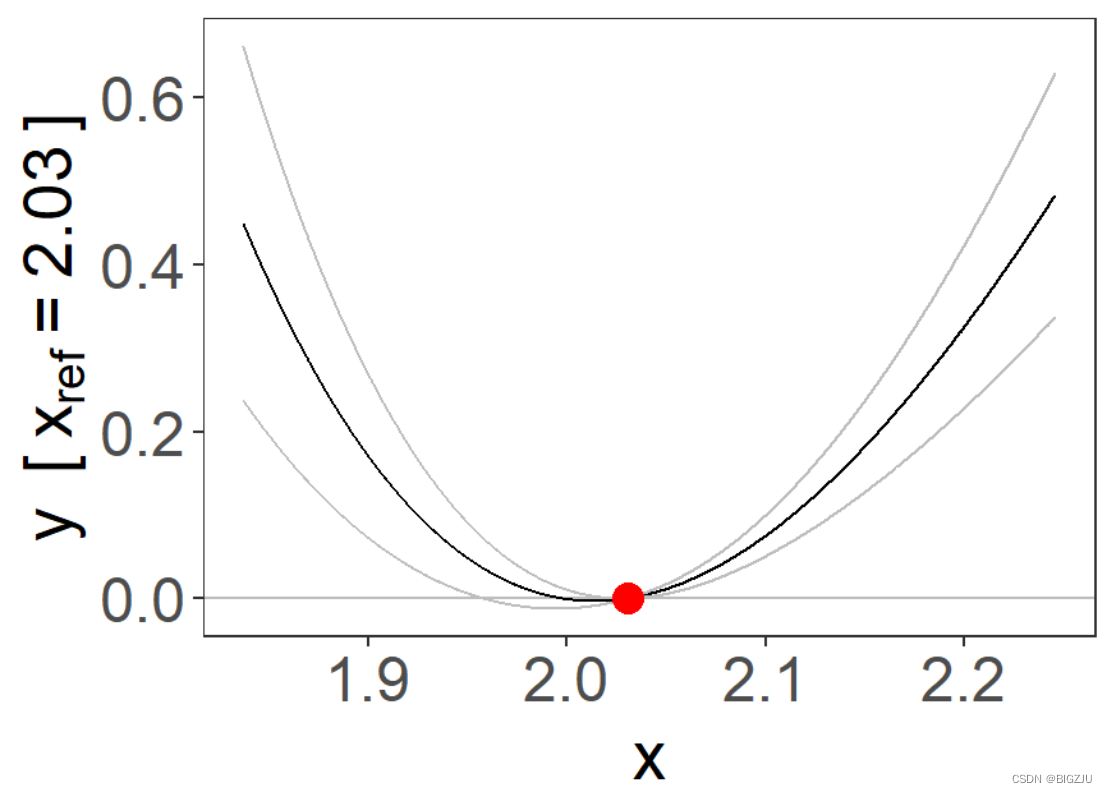
|
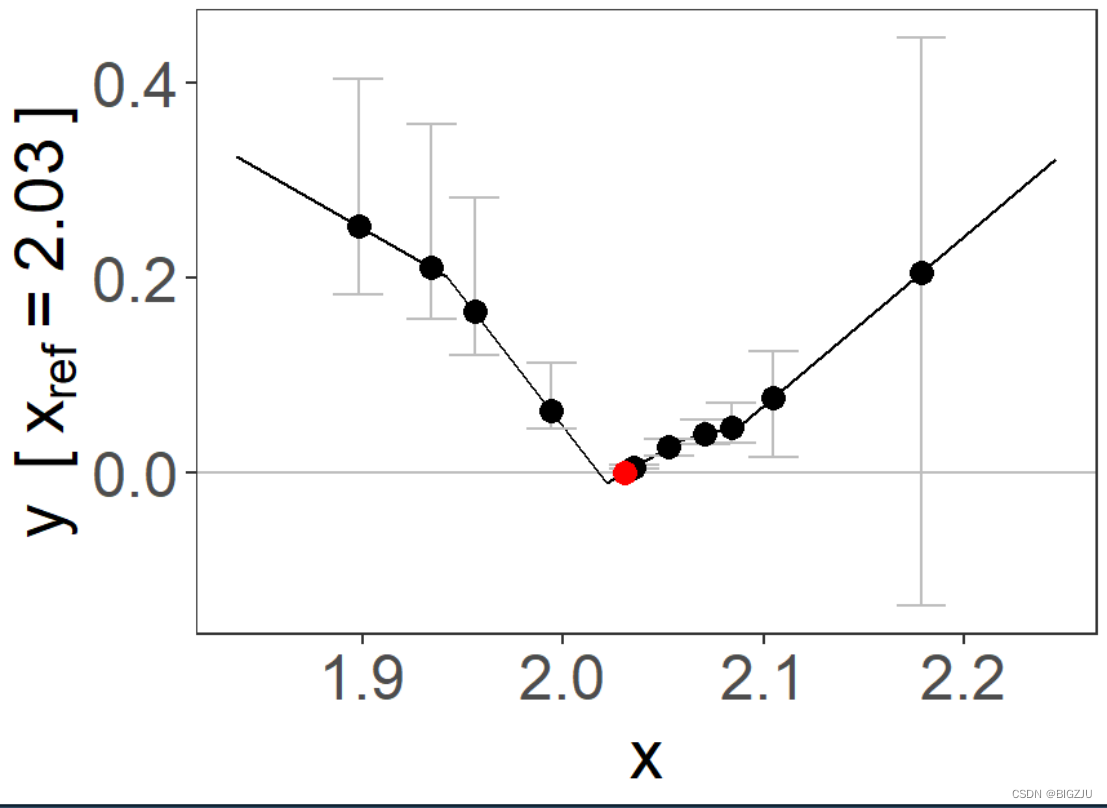
|

















 907
907

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









