



R一些非常用函数
- 1. identical
- 2.surv_cutpoint
- 3.多个数据的交集
- 4.首字母大写
- 5.WGCNA
- 6.创建统计表
- 7.下载kegg所有通路的所有基因
- 8.批量替换
- 9.箱
- 10.添加线以及查看默认ggplot画图的颜色
- 11.导出图为pptx
- 12.jimmy老师的GEO包的快捷查找探针基因方法
- 13.ggplot 我的常用修饰
- 14. 箱线图加蜂巢图
- 15. R查找是否正态变量函数
- 16.t检验与wilcoxon检验
- 17.批量logistics回归
- 18 关于多变量同时循环时
- 19.取消科学计算法显示
- 20.代码变得整洁好看
- 21.当你的复制微信公众号的代码首行有数字或空格时
- 22.查看源代码
- 23.批量提取生存分析
- 24.R语言表达式
- 25 R版本更新后更新R包
- 26 使用bedtools
- 27 相关性
- 28 roc 多条线在一个图上
- 29 多个值替换
- 31 下载kegg通路所有基因
1. identical
identical(x,y) ## 鉴别两个对象是否完全一致,等同于all(x==y)
2.surv_cutpoint
surv_cutpoint,生信分析中生存分析中使用的可以找到合适的划分点使得生存存在差异,
遗憾的是这个函数只能用于两组间的cutoff值,需配合surv_categorize(res.cut)食用
示例:# 0. Load some data
data(myeloma)
head(myeloma)
# 1. Determine the optimal cutpoint of variables
res.cut <- surv_cutpoint(myeloma, time = "time", event = "event",
variables = c("DEPDC1", "WHSC1", "CRIM1"))
summary(res.cut)
# 2. Plot cutpoint for DEPDC1
# palette = "npg" (nature publishing group), see ?ggpubr::ggpar
plot(res.cut, "DEPDC1", palette = "npg")
# 3. Categorize variables
res.cat <- surv_categorize(res.cut)
head(res.cat)
# 4. Fit survival curves and visualize
library("survival")
fit <- survfit(Surv(time, event) ~DEPDC1, data = res.cat)
ggsurvplot(fit, data = res.cat, risk.table = TRUE, conf.int = TRUE)
3.多个数据的交集
con_feature = Reduce(intersect, list(v1 = colnames(table1),
v2 = colnames(table2),
v3 =target$name
))
4.首字母大写
将字母转成大写与小写都太常见了,基本就是toupper那一套,但是首字母大写才是我们比较常见的情形,所以
library(Hmisc)
capitalize(y)
5.WGCNA
自己大概基本整个流程
######### tcga 突变
options(stringsAsFactors = F)
library(maftools)
rm(list = ls())
if (TRUE){
library(survival)
library(survminer)
library(timeROC)
library(data.table)
library(tidyr)
library(dplyr)
library(tibble)
library(caret)
library(limma)
}
pwd = ""
setwd(pwd)
library(readr)
library(dplyr)
library(readxl)
# 数据,临床数据
data_1 = read_xlsx("gse122063_expr..xlsx" ,sheet = 1 ) %>% as.data.frame()
data_2 = read_xlsx("gse37263_expr.xlsx" ,sheet = 1)%>% as.data.frame()
group_1 = read_xlsx("122063分組.xlsx" ,sheet = 1)%>% as.data.frame()
group_2 = read_xlsx("37263分组.xlsx" ,sheet = 1)%>% as.data.frame()
con_gene = intersect(data_1[,1],data_2[,1])
rownames(data_1) = data_1[,1]
rownames(data_2) = data_2[,1]
data_1 = data_1[,-1]
data_2 = data_2[,-1]
data_all = cbind(data_1[con_gene,],data_2[con_gene,])
colnames(group_1) = colnames(group_2)
pheno = rbind(group_1,group_2)
batch = c(rep(1,dim(group_1)[1]),rep(2,dim(group_2)[1]))
modcombat = model.matrix(~1, data=pheno)
edata = data_all
edata = matrix(data = as.matrix(data_all), nrow=4748, ncol = 116)
mode(edata) = "numeric"
combat_edata = ComBat(dat=edata, batch=batch, mod=NULL, par.prior=TRUE, prior.plots=FALSE)
##导入数据##
colnames(combat_edata) = c(group_1$SAMPLE,group_2$SAMPLE)
rownames(combat_edata) = con_gene
library(WGCNA)
options(stringsAsFactors = FALSE)
enableWGCNAThreads()
##筛选方差前25%的基因##
expro = combat_edata
datExpr=as.data.frame(t(combat_edata));
nGenes = ncol(datExpr)
nSamples = nrow(datExpr)
##样本聚类检查离群值##
gsg = goodSamplesGenes(datExpr, verbose = 3);
gsg$allOK
sampleTree = hclust(dist(datExpr), method = "average")
plot(sampleTree, main = "Sample clustering to detect outliers"
, sub="", xlab="")
save(datExpr, file = "FPKM-01-dataInput.RData")
##没有离群值,不作处理##
##软阈值筛选##
powers = c(c(1:10), seq(from = 12, to=20, by=2))
sft = pickSoftThreshold(datExpr, powerVector = powers, verbose = 5)
par(mfrow = c(1,2));
cex1 = 0.9;
plot(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
xlab="Soft Threshold (power)",ylab="Scale Free Topology Model Fit,signed R^2",type="n",
main = paste("Scale independence"));
text(sft$fitIndices[,1], -sign(sft$fitIndices[,3])*sft$fitIndices[,2],
labels=powers,cex=cex1,col="red");
abline(h=0.90,col="red")
plot(sft$fitIndices[,1], sft$fitIndices[,5],
xlab="Soft Threshold (power)",ylab="Mean Connectivity", type="n",
main = paste("Mean connectivity"))
text(sft$fitIndices[,1], sft$fitIndices[,5], labels=powers, cex=cex1,col="red")
##作图##
betal = 10
dev.off()
k.dataOne <- softConnectivity(datExpr, power = betal) -1
scaleFreePlot(k.dataOne, main = paste(" power=", betal))
##一步法网络构建:One-step network construction and module detection##
net = blockwiseModules(datExpr, power = betal, maxBlockSize = 6000,
TOMType = "unsigned", minModuleSize = 30,
reassignThreshold = 0, mergeCutHeight = 0.25,
numericLabels = TRUE, pamRespectsDendro = FALSE,
saveTOMs = TRUE,
saveTOMFileBase = "AS-green-FPKM-TOM",
verbose = 3)
table(net$colors)
##绘画结果展示##
# open a graphics window
#sizeGrWindow(12, 9)
# Convert labels to colors for plotting
mergedColors = labels2colors(net$colors)
# Plot the dendrogram and the module colors underneath
plotDendroAndColors(net$dendrograms[[1]], mergedColors[net$blockGenes[[1]]],
"Module colors",
dendroLabels = FALSE, hang = 0.03,
addGuide = TRUE, guideHang = 0.05)
##结果保存
moduleLabels = net$colors
moduleColors = labels2colors(net$colors)
table(moduleColors)
MEs = net$MEs;
geneTree = net$dendrograms[[1]];
save(MEs, moduleLabels, moduleColors, geneTree,
file = "AS-green-FPKM-02-networkConstruction-auto.RData")
##表型与模块相关性##
trainDt= pheno
rownames(trainDt) = rownames(datExpr)
table(rownames(trainDt) == rownames(datExpr))
trainDt$AD = 0
trainDt$CON = 0
trainDt$AD[trainDt$CLASS=="AD"] = 1
trainDt$CON[trainDt$CLASS=="control"] = 1
moduleLabelsAutomatic = net$colors
moduleColorsAutomatic = labels2colors(moduleLabelsAutomatic)
moduleColorsWW = moduleColorsAutomatic
MEs0 = moduleEigengenes(datExpr, moduleColorsWW)$eigengenes
MEsWW = orderMEs(MEs0)
samples = t(combat_edata)
modTraitCor = cor(MEsWW, trainDt[,c(3,4)], use = "p")
colnames(MEsWW)
modlues = MEsWW
modTraitP = corPvalueStudent(modTraitCor, nSamples)
textMatrix = paste(signif(modTraitCor, 2), "\n(", signif(modTraitP, 1), ")", sep = "")
dim(textMatrix) = dim(modTraitCor)
dev.off()
labeledHeatmap(Matrix = modTraitCor, xLabels = colnames(trainDt[,c(3,4)]), yLabels = names(MEsWW), cex.lab = 0.5, yColorWidth=0.01,
xColorWidth = 0.03,
ySymbols = colnames(modlues), colorLabels = FALSE, colors = blueWhiteRed(50),
textMatrix = textMatrix, setStdMargins = FALSE, cex.text = 0.5, zlim = c(-1,1)
, main = paste("Module-trait relationships"))
## 保存模块与基因
gene = cbind(con_gene,mergedColors)
write.csv(gene,"module_gene.csv")
write.csv(combat_edata,"profile_remove_batch.csv")
6.创建统计表
library(tableone)
data_table = data_exp[,c(2:3,5:13)]
data_table$AGE =ifelse(data_table$AGE>50,">50","<=50")
CreateTableOne(data = data_table)
dput(names(data_table))
myVars <- c("KPS" , "ECOG" , "AGE" , "status" ,
"subtype" , "primarily_detection" ,"GPA" , "naomo" , "number" ,
"danfa" , "neizang" )
catVars <- c("KPS" , "ECOG" , "AGE" , "status" ,
"subtype" , "primarily_detection" ,"GPA" , "naomo" , "number" ,
"danfa" , "neizang" )
tab2 <- CreateTableOne(vars = myVars, data = data_table, factorVars = catVars)
print(tab2,showAllLevels = TRUE)
tab4Mat <- print(tab2,showAllLevels = TRUE, quote = FALSE, noSpaces = TRUE, printToggle = FALSE)
## 保存为 CSV 格式文件
write.csv(tab4Mat, file = "brain_meta_myTable.csv")
7.下载kegg所有通路的所有基因
remotes::install_github("YuLab-SMU/createKEGGdb")
createKEGGdb::create_kegg_db(c('hsa'))
install.packages("KEGG.db_1.0.tar.gz", repos=NULL,type="source")
library(KEGG.db)
library(org.Hs.eg.db)
library(tidyverse)
KEGGPATHID2EXTID <- KEGG.db::KEGGPATHID2EXTID %>%
as.data.frame() %>%
mutate(path_id = str_remove(pathway_id,"hsa"))
KEGGPATHID2NAME <- KEGG.db::KEGGPATHID2NAME %>%
as.data.frame()
KEGG_all <- KEGGPATHID2EXTID %>% left_join(KEGGPATHID2NAME)
# kegg_all里的是基因ID,我需要基因名
kegg <- bitr(unique(KEGG_all$gene_or_orf_id), fromType = "ENTREZID",
toType = c("SYMBOL" ),
OrgDb = org.Hs.eg.db)
KEGG_all = subset(KEGG_all,gene_or_orf_id%in%kegg$ENTREZID)
KEGG_all$gene_name = kegg$SYMBOL[match(KEGG_all$gene_or_orf_id,kegg$ENTREZID)]
# 构造gsva用得上的列表,注意用tapply并不是返回列表
gene_set = KEGG_all
kegg_function<-list()
for(i in unique(KEGG_all$path_name))
{ kegg_function[[i]]<-KEGG_all[i ==KEGG_all$path_name,2]}
8.批量替换
参考链接:https://www.cnblogs.com/ywliao/articles/14535577.html
library(plyr)
x <- c("a", "b", "f")
mapvalues(x, c("a", "b"),c("c", "d"))
9.箱
# 适用文件为基因为列,样本为行的数据。其实想要提醒自己的是,这个组别需为字符类型。
if(T){
data_exp = read.csv("xxx.csv",stringsAsFactors = F, header = T)
dat = data_exp[,2:49] %>% t() %>% as.data.frame()
dat$gene = colnames(data_exp)[2:49]
data_plot=gather(dat,key=group,value=Expression,-gene)
data_plot$group = rep(data_exp$group,each = 48) %>% as.character()
data_plot = na.omit(data_plot)
p = ggplot(data=data_plot,aes(x = gene, y = as.numeric(Expression), fill = group)) +
geom_boxplot(alpha=0.7) +
scale_y_continuous(name = "Expression") +
scale_x_discrete(name = "") +
# ggtitle("Immune Suppressive cytokines") +
theme_bw() +
theme(plot.title = element_text(size = 8, face = "bold"),
text = element_text(size = 12),
axis.title = element_text(face="bold"),
axis.text.x=element_text(size = 11,angle = 90)) +
ylim(0,50) + labs(y="Expression")
m1=p+stat_compare_means(aes(group=group),method ="anova",label = "p.signif",bracket.size = 0.2) #其实默认就是method ="wilcox.test"
m1
10.添加线以及查看默认ggplot画图的颜色
geom_segment加线,ggplot_build(p1)$data这个能看到颜色
# PCOA结果
library(vegan)
df <- dat
bray_dist<-vegdist(df,method = "bray")
library(ape)
df.pcoa<-pcoa(bray_dist,correction = "cailliez")
df.pcoa$vectors
df.pcoa$values
df.plot<-data.frame(df.pcoa$vectors)
head(df.plot)
library(ggplot2)
x_label<-round(df.pcoa$values$Rel_corr_eig[1]*100,2)
y_label<-round(df.pcoa$values$Rel_corr_eig[2]*100,2)
x_label
y_label
df.plot$Group<- data_meta$Group
df.1 = data.frame(df.plot[,1:2] )
df.1$Group = data_meta$Group
# 画正常与疾病的PCOA图
# 确定中点
tapply(df.1$Axis.1,df.1$Group,mean)
tapply(df.1$Axis.2,df.1$Group,mean)
df.1$x = ifelse(df.1$Group=="Autism Spectrum Disorder",-0.00342,0.003709135 )
df.1$y = ifelse(df.1$Group=="Autism Spectrum Disorder",-0.0091,0.0098 )
p = ggplot(data=df.plot,aes(x=Axis.1,y=Axis.2,
color=Group))+
geom_point(size=2)+
theme_bw()+
theme(panel.grid = element_blank())+
geom_vline(xintercept = 0,lty="dashed")+
geom_hline(yintercept = 0,lty="dashed")+
labs(x=paste0("PCoA1 ",x_label,"%"))
p + geom_segment(data=df.1,aes(x=x,y=y,
xend=Axis.1,yend=Axis.2),
arrow = arrow(length=unit(0.001, "cm")),
lineend = "round",
linejoin = "round",
size=0.1)+
annotate("point", x=-0.00342,y=0.003709135,shape=15, color="#00BFC4", size=5) +
annotate("point", x=-0.00342,y=0.003709135,shape=15, color="#F8766D", size=5)
# 画不同批次的箱线图
11.导出图为pptx
library (eoffice)
topptx(画图对象,file_name)
12.jimmy老师的GEO包的快捷查找探针基因方法
https://mp.weixin.qq.com/s?__biz=MzAxMDkxODM1Ng==&mid=2247492027&idx=1&sn=9ae651dda053e3e3778d4557b141b6bd&scene=21#wechat_redirect
https://mp.weixin.qq.com/s?__biz=MzAxMDkxODM1Ng==&mid=2247492037&idx=1&sn=f6a3a38cac4c20b5428f354803444ec4&chksm=9b4ba17eac3c286897898ac8c3cd42418600cf9b206ca3f9c97b04bcf8c49553fcd52ba891ea&scene=178&cur_album_id=1359443730809454593#rd
https://mp.weixin.qq.com/s?__biz=MzAxMDkxODM1Ng==&mid=2247492052&idx=1&sn=e640e0d9468f60616bddf9494117d0b8&chksm=9b4ba16fac3c287934abe0c6515f12af51d018ea2fa9436b2b9b0a1975c5a05318f2178448cd&scene=178&cur_album_id=1359443730809454593#rd
13.ggplot 我的常用修饰
p = ggplot(data2,aes(x=a,y=b))+#指定数据
stat_boxplot(geom = "errorbar", width=0.1,size=0.8)+ #添加误差线,注意位置,放到最后则这条先不会被箱体覆盖
geom_boxplot(aes(fill=a), #绘制箱线图函数
outlier.colour="white",size=0.8,width=0.3)+
### 好的背景,白底无背景线
theme_bw()+
theme(panel.grid.major=element_blank(),panel.grid.minor=element_blank(),
legend.position = 'none',plot.title =element_text
(hjust =0.5),
) +
ylab("Zmax(P2Zmax - P1Zmax)") +xlab("")+
labs(title ="P1-Zmax>=2")+
scale_x_discrete(
labels = c("CAL(-)(n=165)","CAL(+)(n=61)") # 设置y轴刻度
)+ ylim(-3,6)
p <- p + geom_signif(comparisons = list(c("P3-max<2.5","P3-max>=2.5")),#设置需要比较的组
map_signif_level = FALSE,
size=0.8,color="black")
p
tapply(data$Z.基线,data$组别,quantile)
14. 箱线图加蜂巢图
p = ggplot(data2,aes(x=a,y=b))+#指定数据
stat_boxplot(geom = "errorbar", width=0.1,size=0.8)+ #添加误差线,注意位置,放到最后则这条先不会被箱体覆盖
geom_boxplot(aes(fill=a), #绘制箱线图函数
outlier.colour="white",size=0.8,width=0.3)+
### 好的背景,白底无背景线
theme_bw()+
theme(panel.grid.major=element_blank(),panel.grid.minor=element_blank(),
legend.position = 'none',plot.title =element_text
(hjust =0.5),
) +
ylab("Zmax(P2Zmax - P1Zmax)") +xlab("")+
labs(title ="P1-Zmax>=2")+
scale_x_discrete(
labels = c("CAL(-)(n=165)","CAL(+)(n=61)") # 设置y轴刻度
)+ ylim(-3,6)
p <- p + geom_signif(comparisons = list(c("P3-max<2.5","P3-max>=2.5")),#设置需要比较的组
map_signif_level = FALSE,
size=0.8,color="black")
p
tapply(data$Z.基线,data$组别,quantile)
15. R查找是否正态变量函数
seek_normal = function(data,var_need_test,group){
nonvar = NULL
for ( i in var_need_test){
#var_need_test = "住院天数"
num = data[[i]]
#group = data$group
b = tapply(num,group,function(x){shapiro.test(x)})
b = sapply(b,function(x){
x$p.value
})
if (all(b>0.05)){
nonvar = c(nonvar,i)
}
}
return(nonvar)
}
var_need_test = colnames(data_temp)[2:4]
group =data_temp$group_hunhe
var_normal = seek_normal(data_temp,var_need_test,group)
var_nonnormal = setdiff(var_need_test,var_normal)
myVars = colnames(data_temp)[-c(1,5)]
catVars = colnames(data_temp)[c(6,7)]
tab2 <- CreateTableOne(vars = myVars, data = data_temp, factorVars = catVars,strata = "group_hunhe")
tab2
tab4Mat <-print(tab2, nonnormal = var_nonnormal, quote = FALSE, noSpaces = TRUE, printToggle = FALSE,catDigits = 2,contDigits = 2)
tab4Mat
## 保存为 CSV 格式文件
write.csv(tab4Mat, file = "myTable.csv")
16.t检验与wilcoxon检验
####test_two_class对两组连续值做t分布或wilcoxon秩和检验
# data可为数据框或数组,var_normal,var_nonnormal,group分别为正态向量、非正态向量,组别
# 返回值为p值、统计量
test_two_class = function(data,var_normal,var_nonnormal,group){
nonvar = NULL
var_need_test <- c(var_normal,var_nonnormal)
#i = var_need_test[1]
for ( i in var_need_test){
# data=geo_gene
num = cbind(data[,i],group) %>% as.data.frame()
if (i %in% var_normal){
a <- t.test(as.numeric(V1)~group,num)
static <- c(a$p.value,a$statistic)
}
else{
a <- wilcox.test(as.numeric(V1)~group,num)
static <- c(a$p.value,a$statistic)
}
nonvar =cbind(nonvar,static)
}
return(nonvar)
}
17.批量logistics回归
Uni_glm_model<-
function(data,x,group){
#拟合结局和变量
# x = "年龄岁"
data = data
FML<-as.formula(paste0(group,"~",x))
#glm()逻辑回归
glm1<-glm(FML,data=data,family = binomial)
#提取所有回归结果放入glm2中
glm2<-summary(glm1)
#1-计算OR值保留两位小数
OR<-round(exp(coef(glm1)),2)
#2-提取SE
SE<-glm2$coefficients[,2]
#3-计算CI保留两位小数并合并
CI5<-round(exp(coef(glm1)-1.96*SE),2)
CI95<-round(exp(coef(glm1)+1.96*SE),2)
CI<-paste0(CI5,'-',CI95)
#4-提取P值
P<-round(glm2$coefficients[,4],2)
#5-将变量名、OR、CI、P合并为一个表,删去第一行
Uni_glm_model <- data.frame('Characteristics'=x,
'OR' = OR,
'CI' = CI,
'P' = P)[-1,]
#返回循环函数继续上述操作
return(Uni_glm_model)
}
library(plyr)#
#这里我选择了本数据的第2-14列的变量
#把它们放入variable.names中
colnames(data_temp)[9] = "D_D"
variable.names<- colnames(data_temp)[c(1:9)];variable.names
#变量带入循环函数
Uni_glm <- lapply(variable.names,function(x){ Uni_glm_model(data=data_temp,x=x,group="group_hunhe")})
#批量输出结果并合并在一起
library(plyr)#没装的需要先装这个包
Uni_glm<- ldply(Uni_glm,data.frame);Uni_glm
18 关于多变量同时循环时
查看博文:https://www.jianshu.com/p/252a2862adf4
19.取消科学计算法显示
options(scipen = 10)#控制显示位数
20.代码变得整洁好看
加载styler包,然后直接CTRL+A全选代码后,用CTRL+SHIFT+A可自动美化代码
21.当你的复制微信公众号的代码首行有数字或空格时
你可以在你的Rstudio界面,用查找替换功能替换
^\s+\d+ 或 ^\s?\d+ 当空格有多个时用第一个,当没有空格是用第二个。
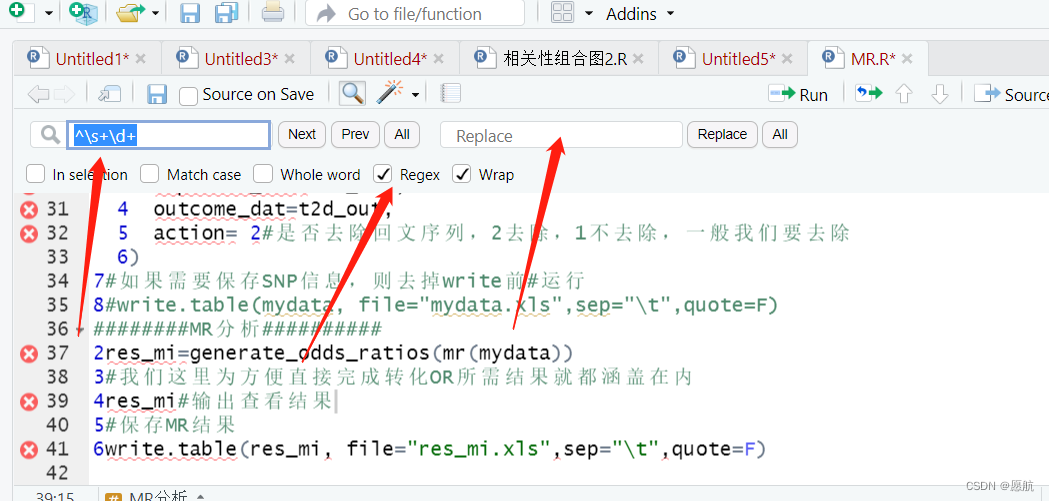
22.查看源代码
stats :::: t.ts)或使用getAnywhere()查看其源代码。
23.批量提取生存分析
covariates <- colnames(df3)[1:308]
univ_formulas <- sapply(covariates,
function(x) as.formula(paste('Surv(OS.time, OS)~', x)))
library(survival)
univ_models <- lapply( univ_formulas, function(x){coxph(x, data = df3)})
# Extract data
univ_results <- lapply(univ_models,
function(x){
x <- summary(x)
p.value<-signif(x$wald["pvalue"], digits=2)
wald.test<-signif(x$wald["test"], digits=2)
beta<-signif(x$coef[1], digits=2);#coeficient beta
HR <-signif(x$coef[2], digits=2);#exp(beta)
HR.confint.lower <- signif(x$conf.int[,"lower .95"], 2)
HR.confint.upper <- signif(x$conf.int[,"upper .95"],2)
HR <- paste0(HR, " (",
HR.confint.lower, "-", HR.confint.upper, ")")
res<-c(beta, HR, wald.test, p.value)
names(res)<-c("beta", "HR (95% CI for HR)", "wald.test",
"p.value")
return(res)
#return(exp(cbind(coef(x),confint(x))))
})
res <- t(as.data.frame(univ_results, check.names = FALSE)) %>% as.data.frame()
p_gene = subset(res,p.value<0.05)
24.R语言表达式
https://www.cnblogs.com/ykit/p/12501664.html


25 R版本更新后更新R包
install.packages("rvcheck")
library(rvcheck)
rvcheck::check_r()
rvcheck::update_all(check_R = FALSE,which = c("CRAN","BioC","github"))
26 使用bedtools
有时真得说生信技能树的思路好用,详见参考:https://cloud.tencent.com/developer/article/1642632,注意bedtools输入的两个文件在染色体、位置起始、终止一致即可。
bedtools intersect -a motif1.bed -b ~/dna/exercise/protein_coding.hg38.position -wa -wb
27 相关性
#### 2.做相关性
library(corrplot)
M = cor(geo_gene)
## 2.1 计算哪些相关显著
# mat : is a matrix of data
# ... : further arguments to pass to the native R cor.test function
cor.mtest <- function(mat, ...) {
mat <- as.matrix(mat)
n <- ncol(mat)
p.mat<- matrix(NA, n, n)
diag(p.mat) <- 0
for (i in 1:(n - 1)) {
for (j in (i + 1):n) {
tmp <- cor.test(mat[, i], mat[, j], ...)
p.mat[i, j] <- p.mat[j, i] <- tmp$p.value
}
}
colnames(p.mat) <- rownames(p.mat) <- colnames(mat)
p.mat
}
p.mat <- cor.mtest(geo_gene)
head(p.mat[, 1:5])
pdf("cor.pdf")
corrplot(M, method="circle", order = 'FPC', type = 'lower', diag = FALSE,
p.mat = p.mat, sig.level = 0.05, insig = "blank")
dev.off()
28 roc 多条线在一个图上
### data 数据框,gene需要做roc的变量,group组别变量的列名
mutiple_auc = function(data,gene,group){
auc_list <- list()
for (i in gene){
data_i = data[,c(group,i)]
colnames(data_i) = c("group","expression")
glm1<-roc(data_i$group,data_i$expression)
auc_list[[i]] <- glm1
}
roc_value <- sapply(auc_list,function(x){
as.numeric(x$auc)
})
tpr <- sapply(auc_list,function(x){
as.numeric(x$sensitivities)
})
fpr = sapply(auc_list,function(x){
1-as.numeric(x$specificities)
})
year = rep(1:length(auc_list),times=sapply(tpr,length))
df_plot <- data.frame(tpr = unlist(tpr),
fpr = unlist(fpr),
year = as.character(year))
labels <- sapply(seq_along(auc_list),function(x){
paste0(gene[x]," AUC: ",round(roc_value[x],3))
})
labels
library(ggplot2)
color <- colors() %>% sample(length(auc_list))
p <- ggplot(df_plot, aes(fpr, tpr, color = year)) +
geom_smooth(se=FALSE, linewidth=1.2)+ # 这就是平滑曲线的关键
geom_abline(slope = 1, intercept = 0, color = "grey10",linetype = 2) +
scale_color_manual(values = color,
name = NULL,
labels = labels
) +
coord_fixed(ratio = 1) +
labs(x = "1 - Specificity", y = "Sensitivity") +
theme_minimal(base_size = 14, base_family = "sans") +
theme(legend.position = c(0.7,0.15),
panel.border = element_rect(fill = NA),
axis.text = element_text(color = "black"))
p
}
group1 <- "group"
p1 <- mutiple_auc(data,gene,group1)
p1
ggsave(filename = "NC rocs.pdf",width = 6,height = 6)
29 多个值替换
library(plyr)
jixian$season <- mapvalues(jixian$month, c("03", "04", "05", "06" ,"07", "08", "09", "10" ,"11", "12" ,"01"),
c("spring", "spring","spring","summer","summer","summer","autumn","autumn","autumn",
"winter","winter"))
library(tableone)
table = CreateTableOne(vars = c("性别", "age", "season"),
factorVars = c("性别","season"), data = jixian)
#将生成的table1存成csv格式
table
table_mat = print(table, quote = FALSE, noSpaces = T, printToggle = FALSE)
write.csv(table_mat, file = "TABLEONE.csv")
#30 基因ID等转换
# 查看可用的属性
attributes <- listAttributes(mart)
print(attributes)
# 查看可用的过滤器
filters <- listFilters(mart)
print(filters)
library(BiocFileCache)
library(biomaRt)
install.packages("httr2")
listMarts()
mart <- useEnsembl(biomart = "ensembl",
dataset = "hsapiens_gene_ensembl")
getBM(attributes = c("affy_hg_u95av2", "hgnc_symbol", "chromosome_name", "band"),
filters = "affy_hg_u95av2",
values = c("1939_at","1503_at","1454_at"),
mart = mart)
# 示例rs号
# 使用Ensembl数据库
mart <- useMart("ENSEMBL_MART_SNP", dataset = "hsapiens_snp")
rs_ids <- c("rs123", "rs456", "rs789")
# 获取对应的基因信息
snp_gene_info <- getBM(
attributes = c("refsnp_id", "ensembl_gene_stable_id", "ensembl_gene_name","associated_gene"),
filters = "snp_filter",
values = rs_ids,
mart = mart
)
# 输出结果
library(clusterProfiler)
library(org.Hs.eg.db)
print(snp_gene_info)
name <- snp_gene_info$ensembl_gene_stable_id
symbol <- bitr(
name,
fromType = "ENSEMBL",
toType = c("SYMBOL"),
OrgDb = org.Hs.eg.db
)
31 下载kegg通路所有基因
hsa_kegg <- clusterProfiler::download_KEGG("hsa")
PATH2ID <- hsa_kegg$KEGGPATHID2EXTID
PATH2NAME <- hsa_kegg$KEGGPATHID2NAME
PATH_ID_NAME <- merge(PATH2ID, PATH2NAME, by="from")
colnames(PATH_ID_NAME) <- c("KEGGID", "ENTREZID", "DESCRPTION")
library(biomaRt)
mart <- useDataset("hsapiens_gene_ensembl", useMart("ensembl"))
entrezgene <- PATH_ID_NAME$ENTREZID
# This step need some time
ENTREZID <- bitr(
entrezgene,
fromType = "ENTREZID",
toType = c("SYMBOL"),
OrgDb = org.Hs.eg.db
)
PATH_ID_NAME <- merge(PATH_ID_NAME, ENTREZID, by= "ENTREZID")
MAPK_PATHWAY = subset(PATH_ID_NAME,KEGGID=="hsa04010")
write.csv(MAPK_PATHWAY, file = "MAPK-PATHWAY.csv")


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









