



生信碱移
图形表示学习(TREE)
来自中国科学院的研究者开发了一个基于转换器的图形表示学习(TREE)框架,着力于癌症基因识别任务中的可解释性和可推广性。
神经网络在解析复杂生物网络或者是多模态数据方面有非常大的优势。先前小编分享了一篇基于变分自编码器(VAE)的泛癌框架 DeepProfile,纯数据库挖掘+简单架构(超低成本)登上了顶刊。
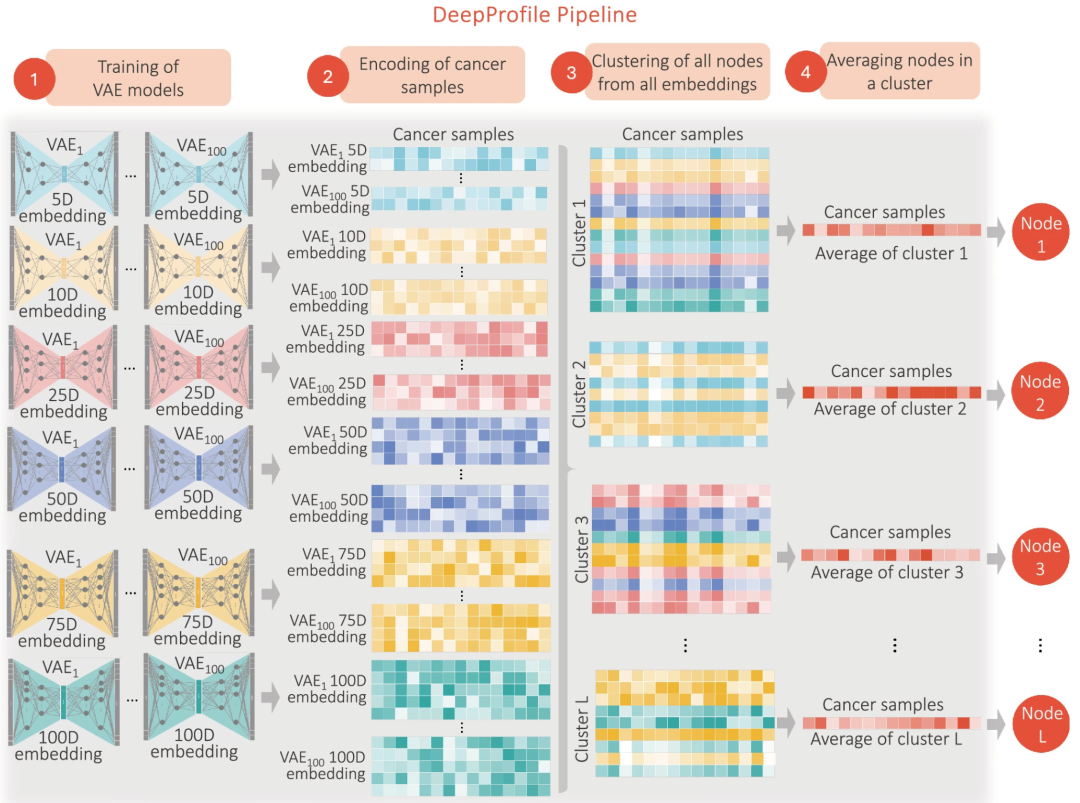
▲ DeepProfile框架:通过训练数百个VAE模型提取癌症样本的潜在变量。每个VAE模型具有不同的潜在维度大小,并进行100次随机权重初始化。使用这些模型对癌症样本编码后,生成多组潜在变量。随后,将所有VAE模型的潜在变量聚类,将具有相似模式的变量组合在一起,不受模型来源或潜在空间大小的约束。最终,通过对每个聚类的变量值进行平均,定义出最终的DeepProfile嵌入和潜在变量,使每个潜在变量成为多个VAE模型变量的集成表示,捕获数据中稳健且一致的生物学特征。
今天小编又看到一篇刚刚见刊的研究,于2025年1月9日发表在Nat. Biomed. Eng [IF:26.8],使用的是 transofomer-based 的模型架构,但是在此基础上针对泛癌多模态的数据输入进行了设计,并且加入了图表示学习的概念。忍不住给各位分享一下!

▲ DOI: 10.1038/s41551-024-01312-5
研究团队
背景
全面了解人类致癌基因是探索肿瘤形成致癌机制的关键基础 。广泛接受的观点认为,累积的基因组改变为细胞提供了选择性生长优势,是癌症形成和进展的根本原因 。这些恶性基因组改变机制包括基因单核苷酸变异(SNVs)、基因拷贝数改变(CNAs)等。
高通量技术的迅速发展为癌症中的基因改变提供了不断扩大的调查,并构建了多个成熟的数据库,如癌症基因组图谱(TCGA)和国际癌症基因组联盟(ICGC) ,以维护大量的人类基因突变数据。受癌症基因倾向于比非癌症基因更频繁突变的观察的启发,通过系统基因组分析已识别出数百个癌症基因。然而,已知癌症基因目录仍然远未完整 。原因有两个方面。① 一方面,并非所有癌症基因的突变频率都很高。一项研究显示,大多数癌症基因的突变频率仅为 2%−20%,这与我们对突变频率的一般认识不符。② 另一方面,癌症的发生是一个复杂的过程,涉及不同类型的基因组改变以及各种分子之间的相互作用,包括但不限于蛋白质、微小 RNA(miRNA)、长非编码 RNA(lncRNA)和转录因子(TFs)。理解多种癌症机制在识别癌症基因中起着关键作用,从而促进癌症的个性化和精准治疗的发展。
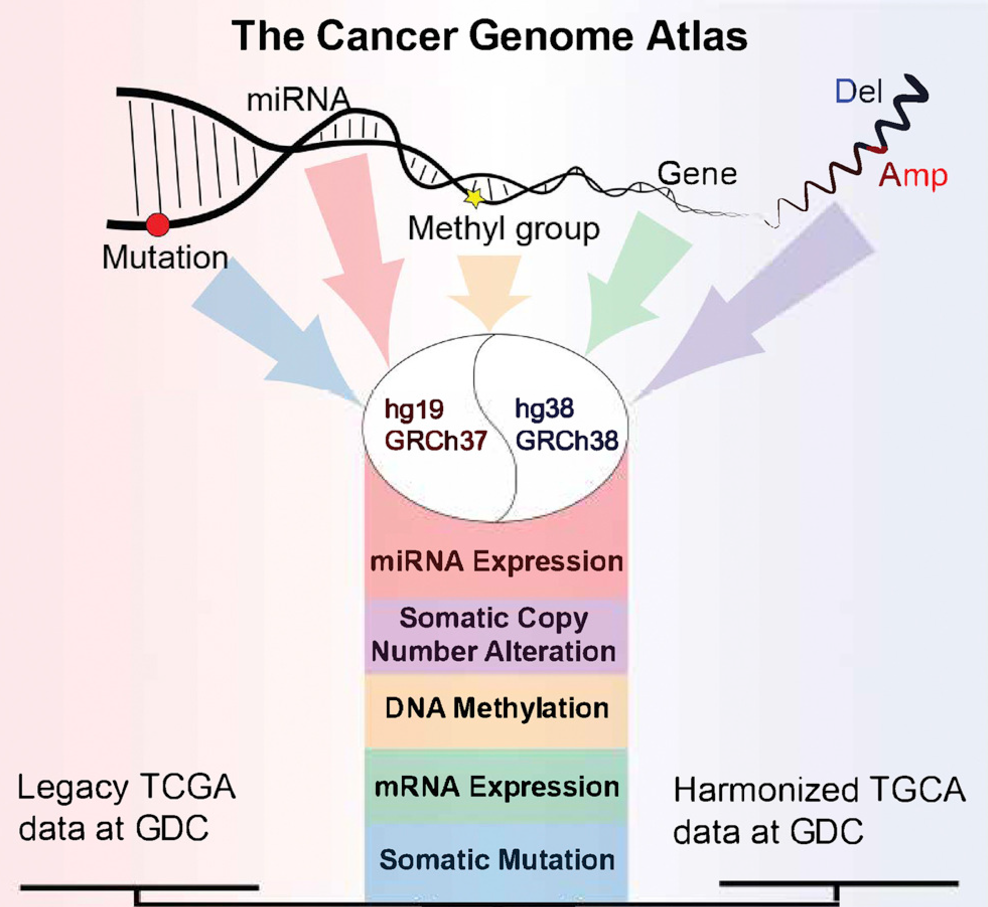
▲ TCGA 数据库存储着多种癌症的多组学数据。from Cell Press
近年来,随着人工智能的发展,癌基因识别的方法学取得了显著进展,这些进展大致可分为基于多组学数据和基于网络的方法。前者通过捕捉不同类型基因组癌症数据中的潜在模式来识别癌基因,但未能考虑基因之间的相互作用。后者基于网络的方法通过提取蛋白质互作(PPI)网络中的特征来区分癌基因,比如近期常常使用的图表示学习(GRL)模型因其强大的表达能力,广泛应用于癌基因的精确识别。尽管这些方法能够整合多组学数据和生物网络信息,但在临床应用中缺乏足够的可解释性和泛化能力。为解决这一问题,本文提出了一种基于 Transformer 的图表示学习框架 TREE,该框架结合了基因的多组学数据和生物网络的拓扑结构,不仅能够提高癌基因识别的可解释性和泛化能力,还能够揭示基因调控的具体机制。TREE通过采样子图和共注意力机制,成功在同质和异质网络中进行癌基因识别,实验结果表明TREE在所有网络上均优于现有的五种网络方法,且能够提供更高的准确性、鲁棒性和可解释性。
方法
输入数据
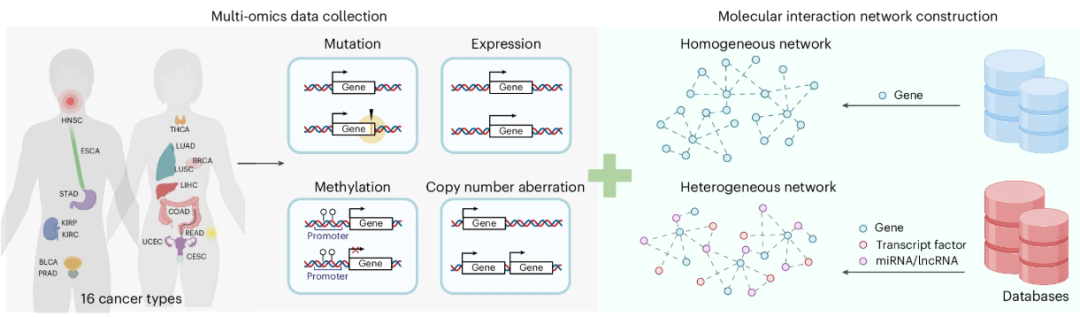
TREE 框架整合了基因突变、基因表达、甲基化和拷贝数异常(CNA)四种组学数据,覆盖 16 种癌症类型。这些数据通过预处理后被作为节点的特征,与构建的生物网络相结合。TREE 使用两种类型的生物网络:同质网络仅包含基因及其蛋白质互作信息,而异质网络进一步引入转录因子(TF)、miRNA 和 lncRNA 等分子节点,捕捉复杂的多分子交互关系。通过局部子图采样,TREE 在保持网络结构信息的同时降低了计算成本。
架构设计
TREE 的架构基于 Transformer,结合局部和全局网络特征进行基因表示学习。节点的多组学特征和网络拓扑位置信息(如节点度和最短路径距离)被嵌入输入层。通过共注意力机制(CA)捕捉异质网络中分子间的全局交互,同时利用多头自注意力机制(SA)聚合来自多子图的特征表示,从而生成目标基因的深度嵌入表示,用于分类是否为癌基因。其具体设计如下:
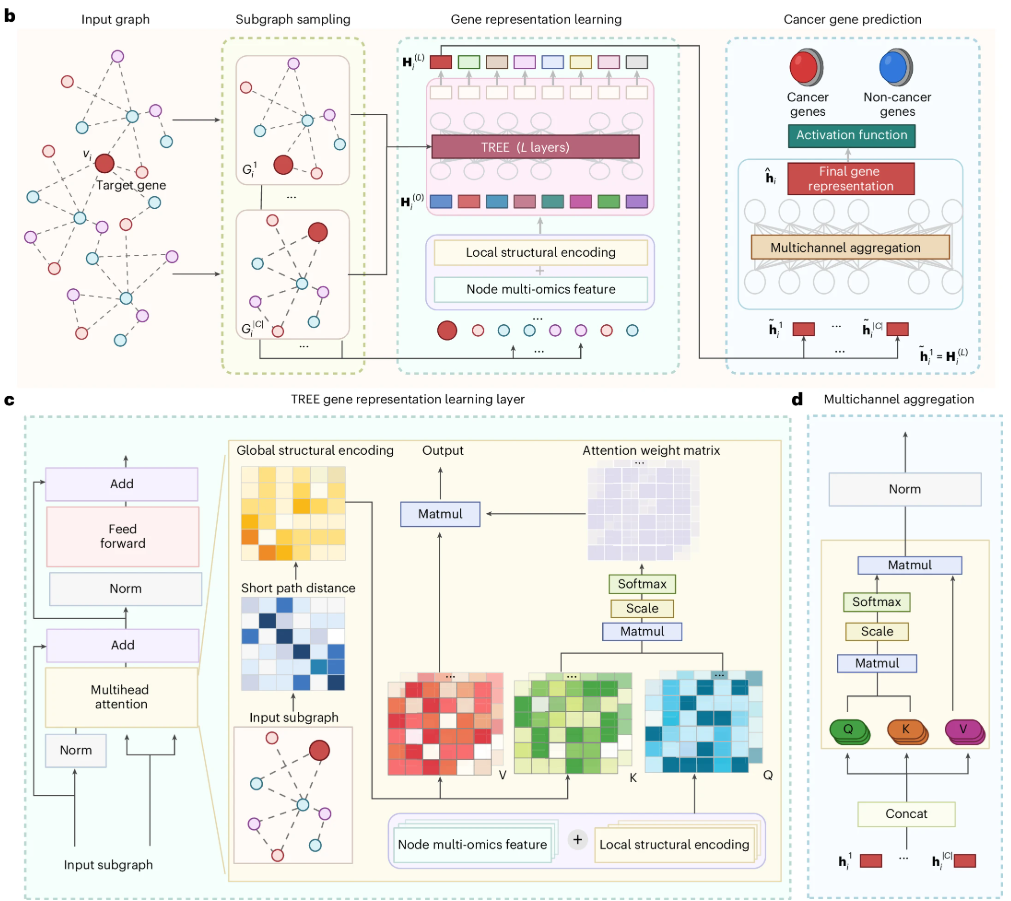
图1:TREE的架构原理。① 输入数据整合与子图采样(图 b 左侧)首先,将多组学数据(如突变、表达、甲基化、拷贝数异常)与分子交互网络(同质或异质网络)结合,构建输入图。以目标基因为中心,采样局部子图 ,这些子图包含目标基因及其邻域的节点和边,捕获了与目标基因相关的局部结构信息,既降低计算开销,又确保重要的交互特征不丢失;② 基因表示学习(图 b 中部):TREE 的核心表示学习模块采用基于 Transformer 的多层结构。每一层整合局部结构特征和全局网络拓扑信息。局部结构编码将节点的多组学特征作为输入,结合目标基因子图中的邻域信息进行处理,生成初步节点表示。全局结构编码(图 c 左侧)使用最短路径距离表示节点间的全局关系,生成节点的全局结构嵌入。这种全局特征通过矩阵计算嵌入到 Transformer 的注意力机制中。多头注意力机制(图 c 右侧)随后将节点的局部和全局特征整合,通过 Query(Q)、Key(K)、Value(V)的矩阵计算生成注意力权重,捕捉重要的交互关系和节点间的相关性。③ 特征聚合与分类(图 b 右侧)多个子图的学习特征经过多通道聚合模块(图 d),利用多头自注意力机制将所有子图的特征表示融合,生成目标基因的最终深度表示。随后,通过激活函数对目标基因进行分类,输出其是否为癌基因的置信分数。
训练目标
TREE 的训练目标是通过节点表示学习优化分类精度,同时捕获多组学和拓扑层面的可解释性。简单来讲,TREE 的目标是对目标基因进行分类,即预测基因是否为癌基因(癌基因或非癌基因)。这是一个节点分类任务,模型的输出为每个基因节点属于“癌基因”类别的置信度(概率值),任务目标是最小化分类误差加权交叉熵损失(Cross-Entropy Loss),提高预测准确性。
其使用的基因标签来自已知的公共数据库:
-
Network of Cancer Genes (NCG):提供已知的癌基因清单。
-
COSMIC(Catalogue of Somatic Mutations in Cancer):记录已知的癌基因及其突变情况。
模型的应用
TREE 同时通过整合多组学数据和基因调控网络来识别与癌症相关的基因,他们的消融研究进一步证实了多组学数据和基因调控网络各自的效用。然后,可以通过回答以下研究问题来评估 TREE 的可解释性:
-
Q1:哪种类型的组学数据对于识别癌症基因更为关键?
-
Q2:哪些网络路径更为关键,并且对识别癌症基因有显著贡献?
-
Q3:哪些图模式能最大化 TREE 的预测能力?
Q1:突变在癌症基因识别中至关重要,而 TREE 在定位罕见突变基因方面具有优势
为阐明四类组学数据的影响,作者利用模型梯度评估其重要性(详见方法部分),结果如图 3a 所示。综合所有癌种来看,突变是区分基因的关键因素。而在癌种特异性情景下,不同组学数据对基因识别的重要性各异。在16种癌种中,8 种以突变或SNVs为主要因素,6 种以基因表达为主,而在 COAD(结肠腺癌)和 READ(直肠腺癌)中,CNA 影响最大。这与生物学发现一致,CNA 是结直肠癌的重要标志物,并可预测患者对贝伐珠单抗联合治疗的预后。
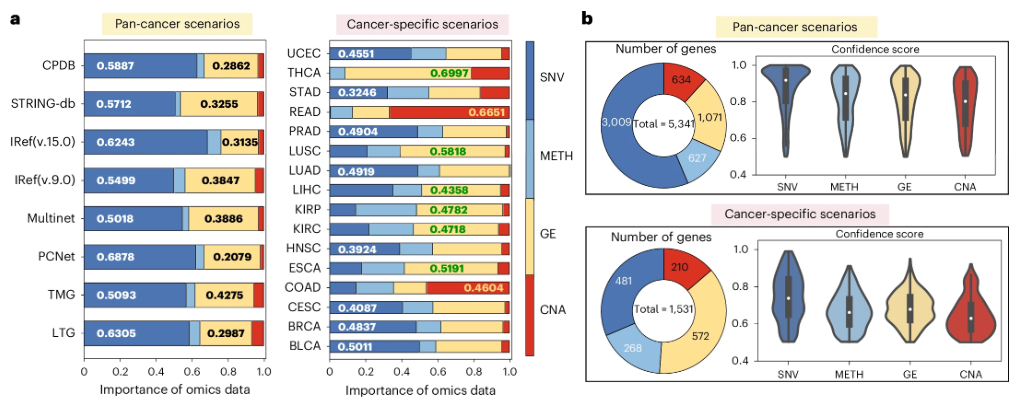
▲ 图3:omics 数据的可解释分析。
Q2:异质信息使 TREE 能够通过网络路径验证癌症基因的重要调控机制
TREE 利用异质网络(如 TF-miRNA-基因网络和 lncRNA-TF-基因网络)识别癌基因,通过网络路径验证其调控机制。异质网络整合了基因-基因、miRNA-基因、TF-基因等多种分子交互信息,为建模不同类型分子间的调控依赖性,定义了7种元路径。这些元路径分别代表可能的功能性调控机制,通过从异质网络中提取子网络实例,TREE 学习其基因嵌入表示并评估性能(图 4a)。结果显示,综合使用所有元路径能显著提高模型在 TF-miRNA-基因(AUPRC 提高 14.31%)和 lncRNA-TF-基因网络(AUPRC 提高 4.345%)上的表现,表明单一调控机制不足以全面识别癌基因。
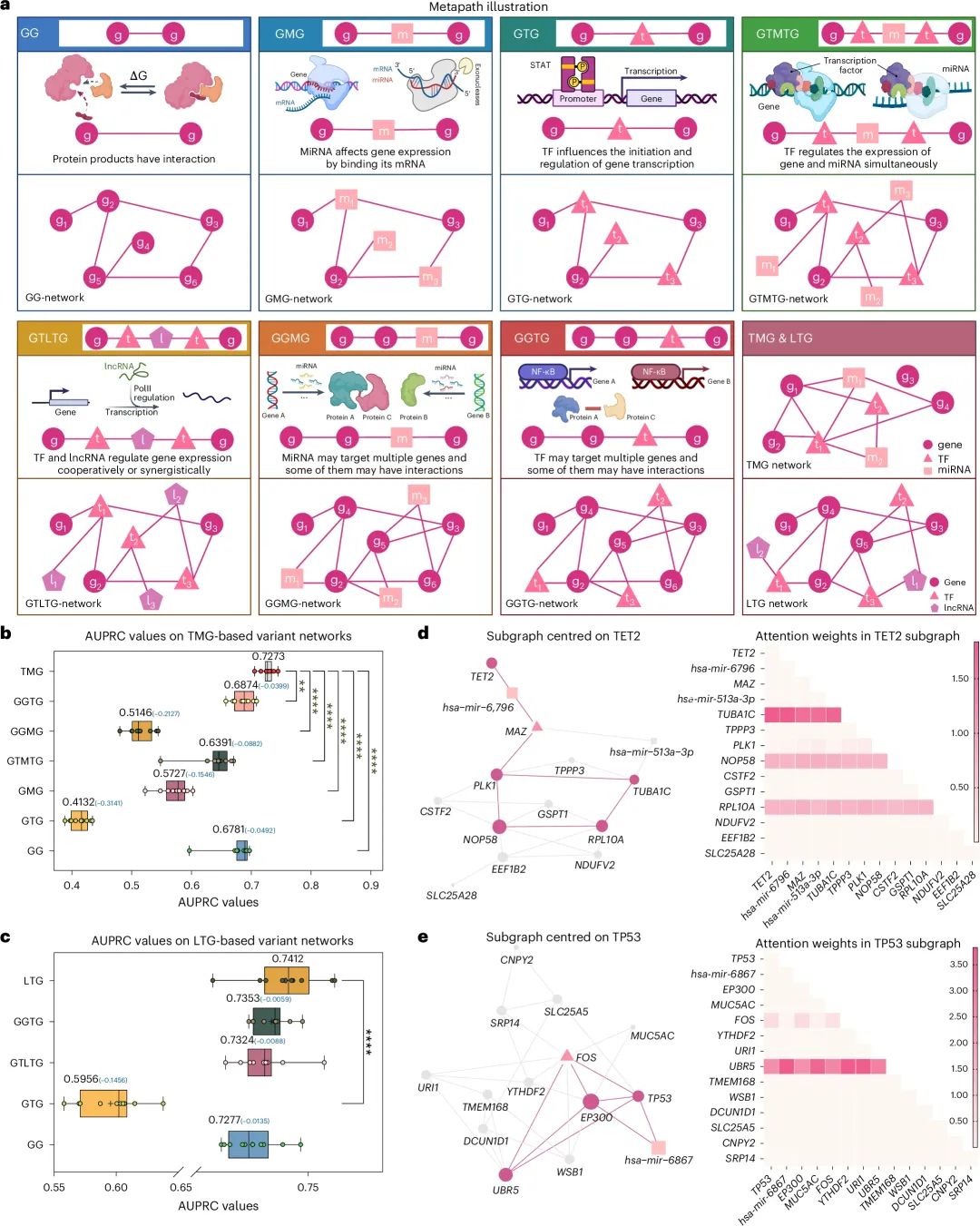
▲ 图4:网络路径上 TREE 的可解释分析。
研究进一步分析了不同元路径的贡献(图 4b, c)。在 TF-miRNA-基因网络中,基因共表达或直接互作的 GG 元路径表现最好,其 AUPRC 比 GTMTG、GMG 和 GTG 分别高出 3.91%、10.54% 和 26.49%,这表明共表达或直接互作的基因更可能驱动癌症的改变;而 GTG 尽管整体表现最差,但能为癌基因和非癌基因的分类提供最高置信分数。在 lncRNA-TF-基因网络中,GTLTG 元路径表现最佳,其强约束条件(如相同调控通路)使其在识别癌基因上更准确。
此外,案例研究进一步展示了 TREE 在复杂调控机制下的有效性。例如,TET2 仅能通过所有元路径的结合正确预测,其网络路径揭示了 hsa-mir-6796 调控 MAZ 和 TET2 的表达,影响细胞周期和肿瘤抑制功能(图 4d)。另外,TP53 则通过与 UBR5 的直接和间接相互作用被识别,UBR5 通过调节 TP53 的泛素化影响其稳定性,解释了其在肿瘤抑制功能中的作用(图 4e)。综上,TREE 利用多样化的元路径和复杂调控机制,有效捕捉了癌基因的潜在调控关系,为新癌基因的发现提供了重要工具。
Q3:最短路径信息赋予了适应性优化基因感受野图结构的能力
TREE 使用最短路径信息建模节点间的全局依赖性,使得在网络结构上不直接相连的节点也能通过 Transformer 的注意力机制有效影响目标基因的预测。例如,对于基因 TP53 的预测(图 5a),TREE 能够捕获其感受野中与其长距离相关的节点(如 CTBP2、KRT18、TLE2 和 PDCD10),这些节点虽然与 TP53 没有直接连接,但它们在模型最后一层中对 TP53 的预测起到了关键作用,验证了长距离依赖在癌基因识别中的重要性。
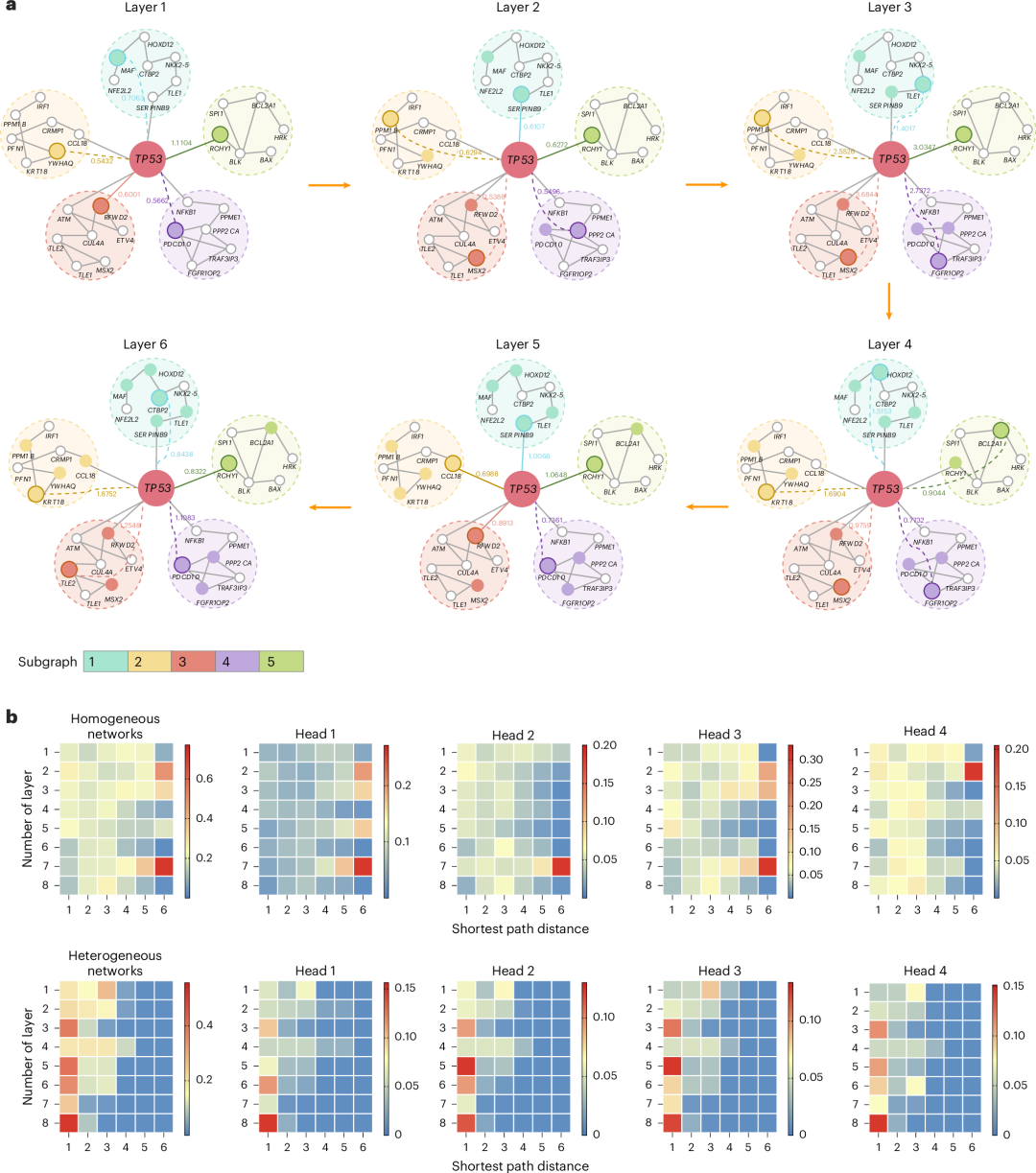
▲ 图5:图形模式的 TREE 可解释分析。
TREE 通过共注意力机制,不再局限于固定的网络结构层级,而是基于注意力权重动态调整每一层的感受野。例如,在 TP53 的预测过程中,第一层中 MAF 基因被识别为关键节点(注意力权重 0.7063),而在第二层,SERPIN89 由于直接与 TP53 相连(权重 0.6107)取代了 MAF 的重要性。随着网络层级的增加,TREE 逐渐关注到距离更远但生物学意义更强的节点(如 TLE1 和 HOXD12),展现了其动态调整感受野的能力。此外,在最后一层中,TREE 将 CTBP2 识别为对 TP53 嵌入学习最关键的节点,进一步印证了感受野优化的有效性。
这种利用最短路径优化感受野的方式与生物学知识高度一致。例如,虽然 TP53 与 MAF 在癌症发展中的关联尚未完全理解,但研究表明 TP53 能够结合 MAF 启动子并抑制其转录活性,从而降低 MAF 的表达水平。同时,MAF 在多种癌症类型中表现过表达,并被认为促进癌症的发展和进程。这些生物学证据支持了 TREE 在决策过程中对 MAF 等节点的重要性判断。
如果泛化到数据多的疾病
又得是篇不错的文章吧
今天就分享到这里了


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









