



 MapBeans是一套基于JavaSwing的地图组件,适用于为Java应用程序添加GIS功能。它包含基础组件Beans,提供地图显示功能,并支持不同数据类型的独立层显示。此外,提供了TableofContents组件用于管理地图层的显示。
MapBeans是一套基于JavaSwing的地图组件,适用于为Java应用程序添加GIS功能。它包含基础组件Beans,提供地图显示功能,并支持不同数据类型的独立层显示。此外,提供了TableofContents组件用于管理地图层的显示。
Map Beans对象:基于Java Swing的一套组件,通常用来添加地图(Map)到Java 2应用程序或小程序的GUI中。
包括:
Beans:基础组件,可以用来添加GIS功能到一个J2SDK应用程序中。所有MO可视化组件工作在JFC框架下。那么我们可以在IDE中载入MO JAR文件,然后就可以用拖/拽的方式来设计应用程序了。
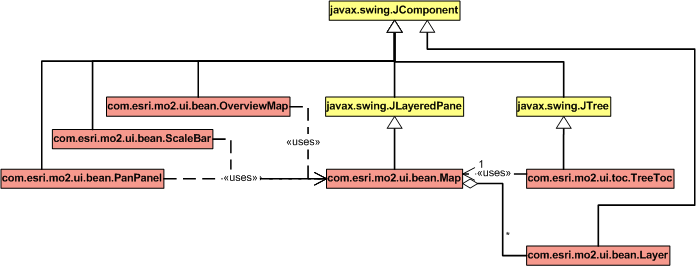
Map:提供一种可视化的途径显示你的空间数据。每种数据类型在一个单独的层(Layer)中显示。你通常从一个层源(LayerSource)。层添加到层集(Layerset)中,然后再添加到地图中。地图显示管理工具可以安排各层按正确绘制顺序显示。地图的setExtent方法是绘制地图的基本途径。地图层集中所有层将响应并绘制它们各自的空间数据。另外一个绘制途径是地图的redraw方法,这个方法简单地绘制图层;不改变地图的区域。如果你的应用程序改变层中某属性(比如:已选择或当前选择选项),使用redraw方法。
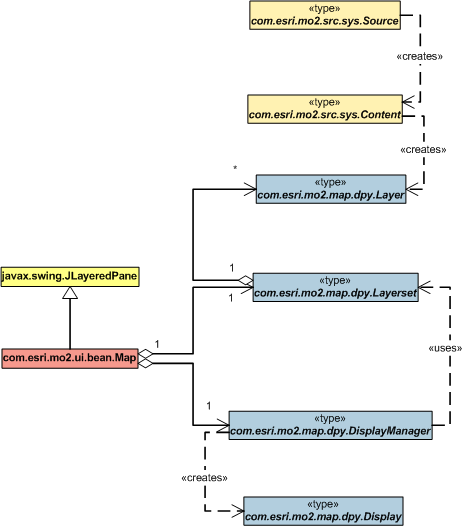
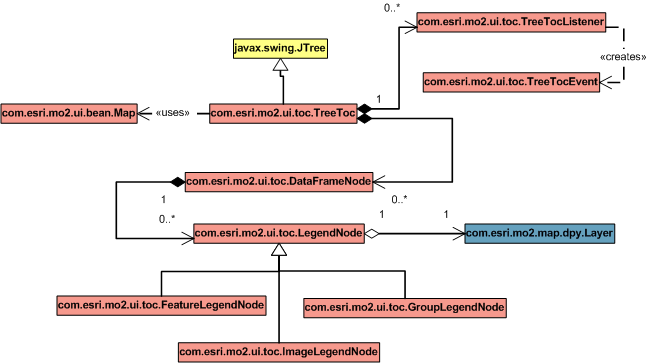
Table of Contents:TreeToc(TOC组件)提供一种地图层显示的途径。每个图例节点对应一个层,包括了层标题和一个复选框,通过复选框控制层的显示。图例节点通常还附带一些信息,比如:颜色和被选等级。使用TOC前你必须用地图的setMap方法将它关联到地图中。你可以点击选择一或多个图例节点,这会高亮显示它们。TOC有三种行为来显示图例节点,用setLegendOutofScaleBehaviour方法设置行为。默认是OUT_OF_SCALE_LEGEND_HIDE(当关联的层不符合规定比例尺,图例节点不可见),另外两个是OUT_OF_SCALE_LEGEND_DISABLE(当关联的层不符合规定比例尺,图例节点不可操作)和OUT_OF_SCALE_LEGEND_DO_NOTHING(当关联的层不符合规定比例尺,图例节点保持可见和可操作)。
AcetateLayers:
Tools
RubberBands
Toolbars
QueryBuilder
Other query dialog boxes
Other dialog boxes
PageLayout

















 664
664

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









