




 本文介绍了基于Reads比对的宏基因组分析方法,包括Kranken2物种注释、HUMAnN功能注释、FMAP功能注释,以及差异和相关性分析。通过对高通量测序数据进行比对,揭示微生物群落的物种组成、功能特征和环境关联。
本文介绍了基于Reads比对的宏基因组分析方法,包括Kranken2物种注释、HUMAnN功能注释、FMAP功能注释,以及差异和相关性分析。通过对高通量测序数据进行比对,揭示微生物群落的物种组成、功能特征和环境关联。
- 介绍
宏基因组 ( Metagenome) 指特定环境下所有生物遗传物质的总和。它包含了可培养的和未可培养的微生物的基因。一般从环境样品中提取基因组DNA, 进行高通量测序,从而分析微生物多样性、种群结构、功能信息、与环境之间的关系等。
宏基因组的分析目前主要包括三种方法:基于组装分析、基于reads分析、基于bin分析。
下面我们介绍基于Reads比对的分析方法。
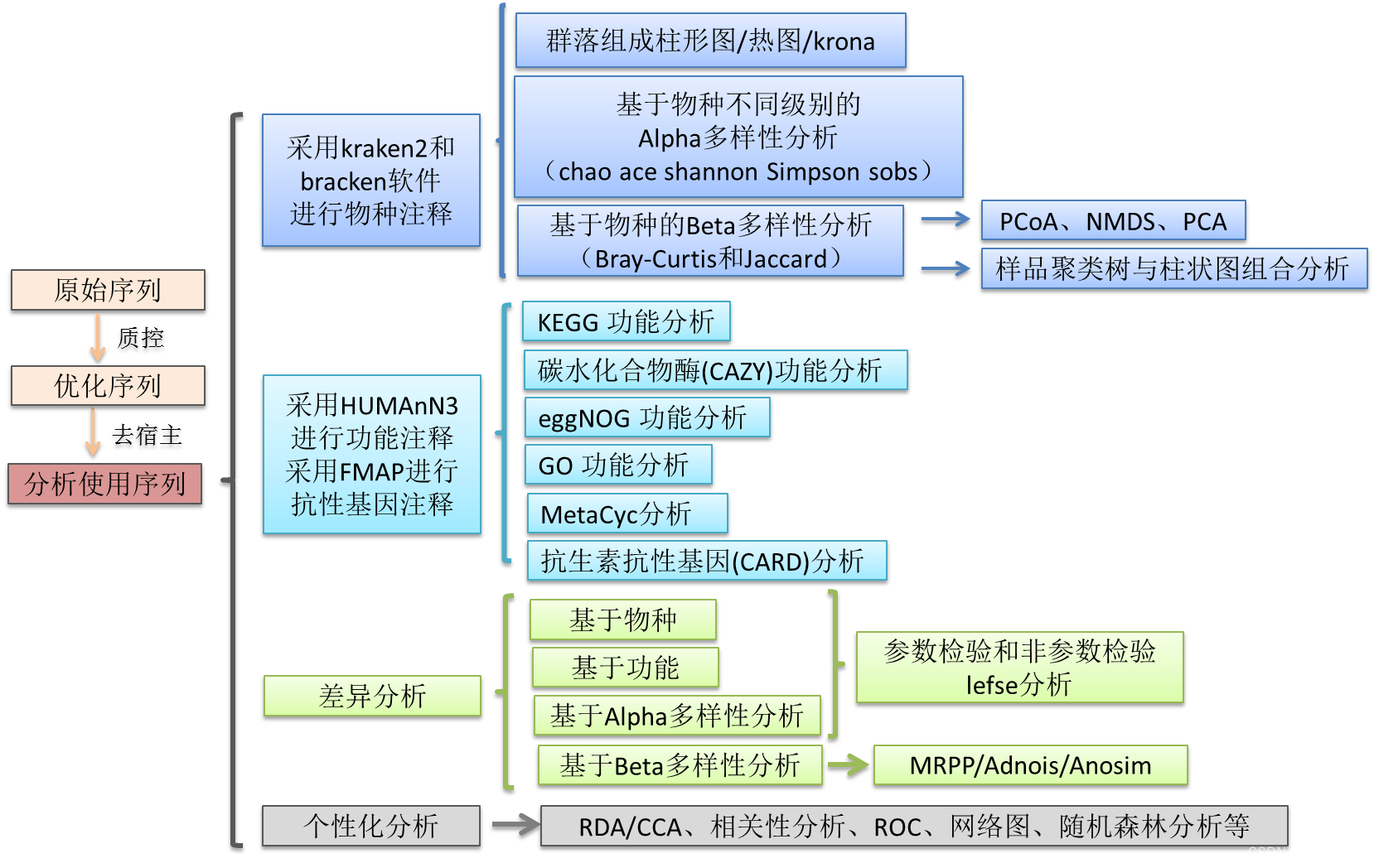
- 分析流程介绍
基于Reads的宏基因组分析,使用质控的优化序列与已知物种和功能数据库进行比对,从而得到每个样本的物种、功能注释信息和相对丰度。此方法的优点能够用比较科学的方法鉴定功能的物种来源,既能研究功能组成,也能研究每个功能都是来自哪些物种;分析速度快。
 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









