



35 电子自旋共振对超高分子量聚乙烯中大自由基的研究
35.1 引言
电子自旋共振(ESR)是一种可直接检测和量化原子或分子体系中未成对或奇数电子的技术。含有未成对电子的材料被称为顺磁性材料,因为它们在外磁场中表现出净磁矩。基于这一原因,ESR也被称为电子顺磁共振(EPR)。尽管这两个名称在实践中均有使用,但已引入第三个名称来替代它们。与核磁共振(NMR)一致,新名称为电子磁共振(EMR),因为ESR/EPR中的磁共振源自电子磁矩。在本章中,使用术语ESR。
电子自旋共振是唯一一种特别适用于直接检测以及对固体或液体中自由基进行定量分析的技术。在聚合物中,自由基通常由于通过机械或化学方式或通过电离辐射辐照导致特定分子位点(主链、侧链或环)上的电子未成对而形成。由于具有未成对电子,自由基成为电子自旋共振研究的良好候选对象。20世纪50年代中期电子自旋共振仪器问世后不久,便开始了包括聚乙烯(PE)在内的聚合物中自由基的测量。有关聚乙烯自由基的大多数早期工作由马尔科姆·多尔汇编成两卷本,书名为《大分子的辐射化学》[1]。读者可参考第一卷的第9、13和14章,以及第二卷的第1、13–16章。1995年,Jan F. Ra‐bek也出版了大量关于聚合物降解的研究著作,其中包括电子自旋共振结果[2]。在后一部著作中,应特别关注第3章和第10章,因为它们与本章主题密切相关。
在低密度聚乙烯(LDPE)中,自由基在室温下不稳定。这种低密度聚乙烯,无论是线性或拉伸的,都是Dole和Rabek前述研究中的主要材料。因此,这些早期研究中的电子自旋共振测量是在液氮温度下进行的。然而—尽管如此,这些对相对简单的聚乙烯的测量结果有助于理解更复杂的超高分子量聚乙烯(UHMWPE)基质中的大自由基过程。
超高分子量聚乙烯(UHMWPE)作为全髋关节和膝关节部件的首选材料,在骨科科学与技术领域已具有五十多年的重要性。然而,由于辐射灭菌或交联过程中在聚合物基体中产生的自由基,UHMWPE的物理和化学性质会受到不利影响[3–13]。因此,电子自旋共振(ESR)技术成为检测辐照后UHMWPE的重要诊断/研究工具。考虑到电子自旋共振(ESR)知识对骨科或生物材料领域具有重要意义,本章从电子自旋共振的基本原理开始,提供了关于在超高分子量聚乙烯(UHMWPE)中产生、传播和终止的自由基的电子自旋共振数据。重点介绍了短期和长期氧化的结果,以及对过氧和氧诱导自由基(OIRs)的鉴定。同时还展示了维生素E掺杂的超高分子量聚乙烯的电子自旋共振结果以及定量电子自旋共振分析。
35.2 电子自旋共振基本原理
电子自旋共振光谱基于电子的基本性质,包括质量($ m_e = 9.11 \times 10^{-31} $ kg)、电荷($ e = -1.6 \times 10^{-19} $ 库仑)和自旋(s)。因此,原子或分子中的未成对电子(例如氢原子、过渡金属离子、自由基)具有磁矩和角动量。磁矩与角动量之比称为旋磁比。由于材料(固体、液体或气体)中电子自旋的随机性,材料的净磁矩可能为零。在外加稳态磁场(H)中,自旋磁矩会平行或反平行于磁场方向排列,电子能级分裂为高(+)和低(−)能态。两能态之间的能量差∆E称为塞曼能,由∆E = gβH给出,其中H为外加稳态磁场,β为电子玻尔磁子(β = 9.27 × 10⁻²⁴ J/T),g为谱分裂因子,通常称为g值。当且仅当振荡磁场的频率f对应的能量等于∆E时,即hf = ∆E = gβH,才能激发这些能级之间的跃迁,其中h为普朗克常数(h = 6.63 × 10⁻³⁴ Js)。条件hf = gβHr被称为共振条件,其中Hr为共振时的外磁场。g值与旋磁比(γ)的关系为 γ = gβ/h,其中β为之前给出的电子玻尔磁子,h为普朗克常数。自由电子的g值为2.0023。自由基种类的g值可用作鉴定机制以确定自由基类型。电子自旋共振的共振条件类似于众所周知的核磁共振,其中g被gN(核g值)取代,β被βN(核磁子)取代。尽管电子自旋共振工作在微波频率(吉赫兹),核磁共振则工作在射频(兆赫兹)。
因此,g值表示为
$$ g = \frac{hf}{\beta H_r} $$ (1)
以及共振磁场位置
$$ H_r = \frac{hf}{g\beta} $$ (2)
对于电子而言,频率f位于微波区域。由于g是某种物种(例如自由基)的常数或特征值,共振场与微波频率成正比。电子自旋共振波谱仪在频率范围∼9 × 10⁹ Hz,即9 GHz下工作,被称为X波段波谱仪;它最常用于研究有机或高分子材料,包括生物材料。有机或高分子自由基的g值仅略偏离自由电子的g = 2.0023。对应于g = 2.0的磁场为3000高斯(G)或0.3特斯拉(T)。核磁共振信号强度是在恒定磁场下作为频率的函数被记录的。与核磁共振波谱仪不同,电子自旋共振波谱仪需要一个在固定频率下工作的谐振器(微波腔),并记录作为外加稳态磁场函数的共振吸收信号。L波段(f = 1 GHz)波谱仪的共振磁场为357 G,S波段(f = 4 GHz)为1430 G,K波段(f = 24 GHz)为8570 G,Q波段(f = 35 GHz)为12,500 G。还有一些其他专用的电子自旋共振波谱仪工作在非常低或非常高的频率下。
在共振时,低能态(−)自旋吸收微波能量产生一个吸收信号通常非常微弱,可能无法直接记录或显示。因此,与核磁共振(NMR)、傅里叶变换红外光谱(FTIR)或光学光谱仪不同,电子自旋共振波谱仪通过在X波段以100 kHz调制外磁场,记录吸收信号的一阶导数。波谱仪也可能具备生成二阶导数吸收信号的选项。例如,分子中因断裂的共价键而形成的未成对电子会产生一个自由基(按定义),只要没有净核磁矩与未成对电子耦合,或不存在可分辨的超精细耦合,电子自旋共振最有可能产生一条单一谱线,即单重态(见图35.1)。图35.1A显示了未成对电子在外加稳态磁场H中的能级状态,其能级分裂为∆E = gβH。(+)和(−)态之间的跃迁在共振时表现为吸收信号的一阶导数,此时∆E = gβHr = hf,其中Hr为共振磁场,f为共振频率。图35.1B展示了一个稳定的有机自由基——2,2‐二苯基‐1‐苦基肼(二苯基苦基肼)所产生的实验测得的电子自旋共振谱。对应的吸收曲线如图35.1C所示,该曲线通过对一阶导数谱线或谱图进行积分获得;曲线下方的阴影区域与自由基浓度(RC)成正比。在计算上,共振磁场位置Hr(即一阶导数曲线的交越点或吸收曲线的峰顶位置)Hr = 3515.824 G,以及线宽∆Hpp = 1.33 G可通过放大实验谱直观测量。如图35.1C所示,峰的磁场位置等于Hr值。共振频率在测量时由仪器自动记录(Bruker EMX)。利用共振公式(1) g = hν/βHr中的频率f = 9.859108 GHz,计算得到g值为g = 2.003544。通过取五次测量的平均值获得了更精确的g值,计算得到的g值为g = 2.00360 ± 0.00005,与教材中的2.0036 ± 0.0006一致。DPPH常被用作定量电子自旋共振或g标记的标准。吸收曲线下方的阴影区域与自由基浓度RC成正比。
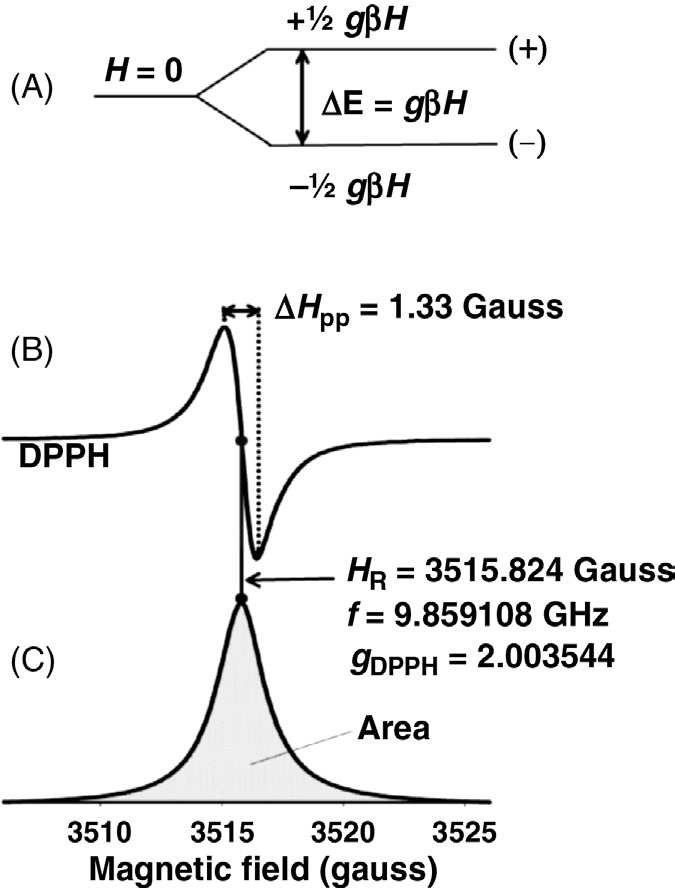
尽管可能无法从单重态中获得自由基的结构信息,但电子自旋共振参数,如谱分裂因子值以及线宽或峰‐峰间距∆Hpp,可以提供关于该自由基的基本信息。关于单重态的进一步讨论将在后文展开。
如果一个质子位于未成对电子(分子断裂位置)附近,每个电子塞曼能级将分裂为两个(对于核自旋I = 1⁄2)或三个(对于核自旋I = 1)能级。对于每个核自旋,电子自旋能级分裂为2I + 1个能级。当核自旋I = 1⁄2时,共振线的磁场位置由下式给出
$$ H_r = H \pm \frac{1}{2}a $$ (3)
其中,Hr是自由基的共振场位置,“a”为超精细耦合常数或超精细常数。在此情况下,得到两条谱线:一条位于H₁ = Hr − 1⁄2 a;另一条位于H₂ = Hr + 1⁄2a。因此,两条谱线之间的间距(H₂ − H₁ = (Hr + 1⁄2 a) − (Hr − 1⁄2 a) = a)为“a”。在多重线谱中,可通过测量超精细分裂或简单地测量谱线间距来确定超精细常数。对于
对于一个邻近含有五个等价质子的自由基,每个I = 1⁄2,在其电子自旋共振谱中观察到六条共振线(2 × 5/2 + 1 = 6);超高分子量聚乙烯中烷基自由基的六重线谱就是一个很好的例子(见图35.2)。在非均相聚合物基质中,由于分子的随机取向,烷基自由基(‐CH₂,b‐ ●CH a‐CH₂,b‐)中的一个a‐质子和四个b‐质子实际上表现为五个等价质子。谱线的强度比(理论值)为1:5:10:10:5:1。由于边峰(或双重峰)的重叠,图35.2中一阶导数谱线的强度以及谱线之间的间距无法提供准确的结果。然而,对于g值的计算,记录了第一条谱线(H = 3296 G)、第六条谱线(H = 3447 G)以及共振中心Hr = 3372 G的磁场位置。利用公式(3)中的工作频率f = 9.4465吉赫兹,计算得到该自由基的g值为g = 2.0017。
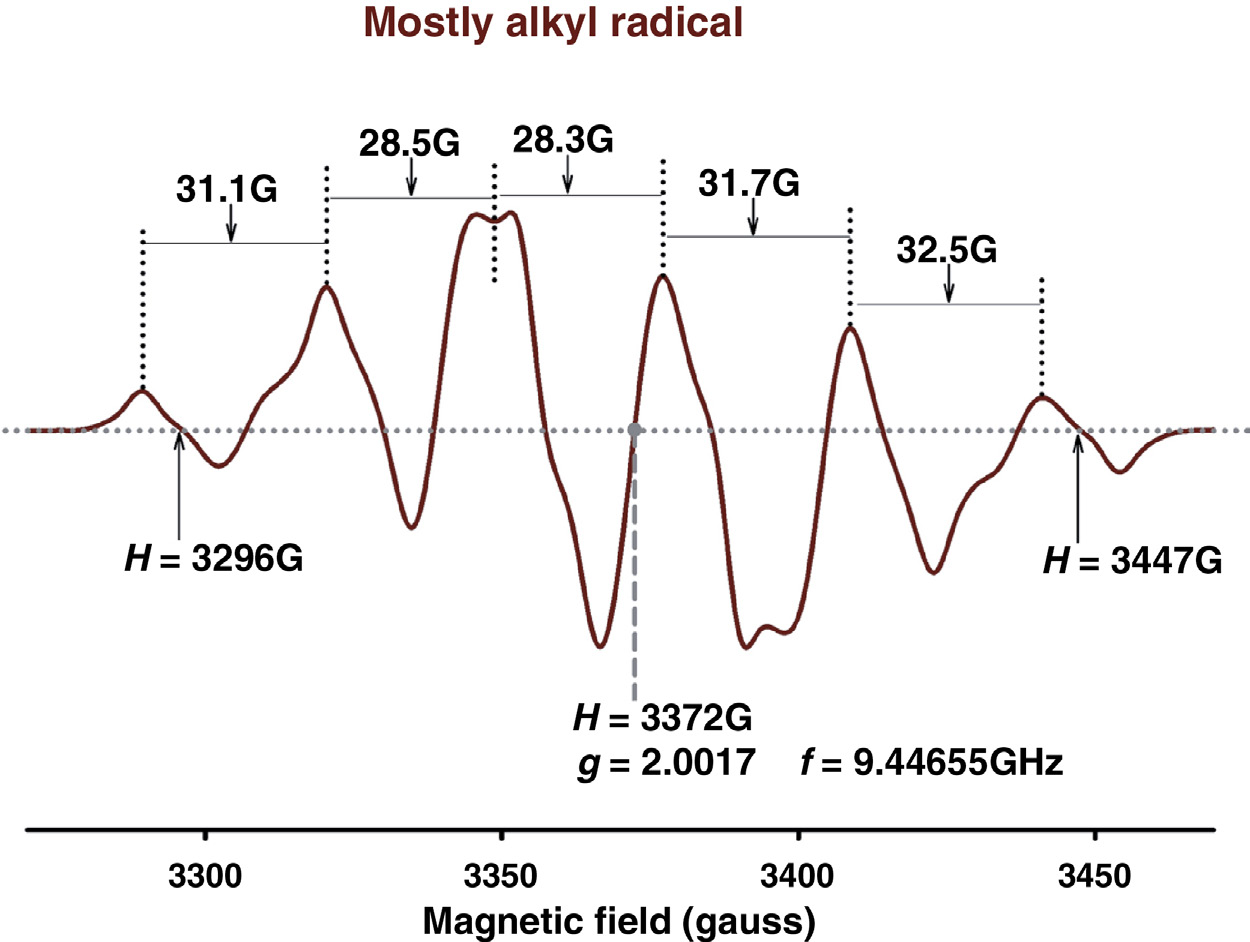
大约。通过测量谱线间距,得到超精细常数的近似值为aalkyl = 30.4 G。然而,如果核自旋I = 1⁄2或1的非等价或不同的质子与同一自由基中的未成对电子发生耦合,则由于不同超精细耦合的组合或重叠效应,谱线位置和/或强度比可能会变得复杂。关于电子自旋共振的背景知识,读者可参考张[14]所著第6章《光谱学基本原理》以及Weil等人的第1章《电子顺磁共振》[15]。
35.3 超高分子量聚乙烯中的自由基
文献中经常报道的主要或初级自由基包括:烷基,‐CH₂‐ ●CH‐CH₂‐;烯丙基,‐CH₂‐CH = CH‐●CH‐CH₂‐或‐CH₂‐ ●CH‐CH = CH‐CH₂‐;以及多烯基,‐HC●‐H(CH = CH)n‐。二级或三级自由基包括:过氧基,‐CHO₂●‐;烷氧基,‐CHO●‐;乙烯基,>CH = ●CH₂‐;以及氧化诱导自由基(OIR)。简而言之,主聚合物自由基表示为P●或R●,过氧自由基表示为PO₂●、POO●、RO₂●或ROO●,烷氧自由基表示为PO●或RO●。下文将对每种自由基进行简要讨论。注意,分子断裂处存在未成对电子通常用上标点符号(●)表示,例如‐●CH‐。
35.3.1 超高分子量聚乙烯中的烷基自由基
烷基自由基是由于从聚乙烯链的‐CH₂‐基团中抽取一个氢原子而形成的。因此,分子或基团(例如‐●CH a ‐)中少一个氢的位置就具有一个未成对电子。在未成对电子位置上的氢原子被称为a‐氢,其最近邻的四个氢原子(左右各两个)被称为b‐氢原子。烷基自由基的分子结构表示为‐CH₂, b‐●CHa‐CH₂, b‐。这些b‐氢原子就它们与未成对电子的相互作用而言被视为等价原子。当然,每个b‐氢原子相对于分子链的取向差异可能使其成为非等价原子。聚乙烯链中‐CH₂‐基团的次近邻氢原子被称为g‐氢原子,它们也可能对电子自旋共振信号产生影响。如前所述,一个未成对电子在电子自旋共振信号(或电子自旋共振谱)中产生一条单共振线。该单共振线分裂为两条等强度谱线(称为超精细线),当其与附近的未成对质子(如氢核)发生耦合时,分裂间距为“a”(称为超精细分裂或耦合常数)。如果同一未成对电子还与其他邻近的质子(例如,b‐氢原子在聚乙烯链中)发生耦合,则超精细线可能进一步分裂,具有不同的耦合常数。然而,在超高分子量聚乙烯中,由于聚合物基体内的分子链呈随机取向,无法区分a‐氢和b‐氢原子对电子自旋共振谱的影响。烷基自由基的电子自旋共振谱中典型的六重线模式源于五个氢原子的平均耦合,其中一个a‐氢和四个b‐氢原子(见图35.2)。谱线间距“a”在23 G到30 G之间变化。图35.2中的每条谱线都较宽,并表现出额外特征,如侧线或卫星线,或双峰。这些谱中的附加特征可能源于超高分子量聚乙烯的复杂性质,因其在任意基体中均含有结晶区、非晶区和界面区域。电子自旋共振谱中的附加特征也可能由不同操作参数引起,例如微波功率、调制幅度、温度、时间常数等。因此,将超高分子量聚乙烯中的电子自旋共振信号归因于某一特定自由基种类变得非常困难。在将信号或谱归属给某一特定种类之前,必须仔细考虑所有这些变量。早在20世纪60年代,许多研究者对辐照聚乙烯进行了非常深入的研究,并基于电子自旋共振谱对每种自由基种类做出了精确的结构归属。例如,多尔(见第14,[2]章)在77 K下对拉伸聚乙烯样品中取向分子的烷基自由基观察到五条超精细线,其间距为28 G(a_b = 28 G),这是由四个等价的b‐氢原子引起的。此外,每条超精细线又被发现分裂为两条,分裂间距为13 G(a_a = 13 G),这是由一个a‐氢原子产生的。
35.3.2 超高分子量聚乙烯中的烯丙基自由基
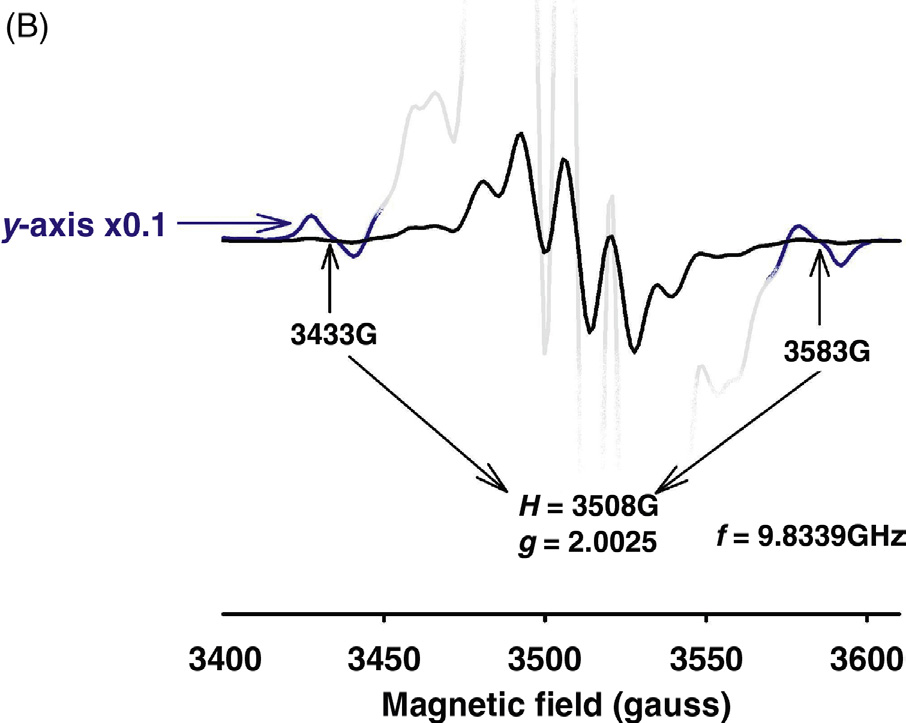
如图35.3所示,为烯丙基自由基‐CH₂-CH= CH-●CH-CH₂- ↔ -CH₂-●CH-CH= CH-CH₂‐的近似分子结构图。如前所述,对聚乙烯自由基的精确结构分析早在20世纪60年代就已开展(见第14,[2]章)。例如,通过使用取向线性聚乙烯在77–293 K温度范围内的实验,大西鉴定出一种由于四个b‐氢和两个a‐氢原子引起的七重线电子自旋共振谱[16,17]。谱线间距约为21高斯。还测量了C1-Ha和C3-Hb键角(见图35.3)对电子自旋共振谱在不同温度下的影响。低温下烯丙基自由基的浓度通过减去该温度下的烷基RC,或在室温下当烷基自由基已衰变后的值来确定。在氮气或真空中于室温进行γ辐照后,超高分子量聚乙烯中可在室温下获得清晰的烯丙基自由基七重线(ESR)指纹。图35.4A显示了该自由基的一阶导数电子自旋共振谱,其超精细耦合常数aallyl ≈ 13.6 G,谱分裂因子g值为g ≈ 2.0016,大约如此。注意,该自由基的g值对应于整个谱的共振磁场(Hr = 3372 G),即中间谱线的交叉点。如果同时存在烷基自由基,则谱图两端(左右两侧)将出现边峰。弱宽峰分别位于左侧和右侧,如放大y轴比例尺,在图35.4B中,六重线谱的成员对应于烷基自由基;其余四条线被烯丙基自由基的强线所覆盖。烷基自由基的中心或共振磁场(Hr = 3508 G)可通过测量两条谱线的磁场位置(3433高斯和3583高斯)近似计算得到。该自由基的g值计算为2.0025,略大于从以烷基为主的电子自旋共振谱(g值,g = 2.0017)获得的结果(图35.2)。尽管每种自由基都有其特征性的a和g值,但此类差异可能由实验误差引起。因此在实际中,对于烷基、烯丙基和多烯基自由基,任何介于2.001和2.003之间的g值均可接受。然而,为了获得统计显著结果,需要进行大量测量并与理论值进行比较。
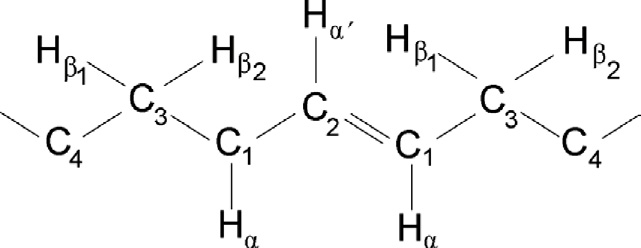
此时有两个重要观察:(1)此类谱可作为烷基和烯丙基自由基同时存在的良好指示剂;(2)通过追踪最右侧或最左侧的其中一条谱线以及中间一条谱线(来自烯丙基)的变化,可以追踪一种自由基在另一种自由基存在下的反应过程。也可采用差减法来确定烷基和烯丙基自由基的浓度;但同时也应考虑二烯基、三烯基、多烯基或乙烯基自由基的存在。为了准确估算组分自由基,可通过应用模拟方法对实验谱进行解卷积(参见第35.3.3节)。
35.3.3 超高分子量聚乙烯中的多烯自由基
大西等人对多烯基自由基CH₂‐●CH(‐CH= CH)n‐ CH₂‐[17],进行了详细讨论,这些自由基是由于烷基自由基迁移而产生的。由于共轭双键的形成,多烯基自由基相当稳定。根据大西等人的研究,多烯基自由基在高剂量(>1000 kGy)下产生,而烯丙基自由基在中等剂量(100–1000 kGy)下形成,烷基自由基则在低剂量(<10 kGy)下产生。然而,在低剂量下也会生成少量的多烯基自由基,但由于其他自由基具有非常相似的g值并存在强烈的共振线,可能无法直接观察到。
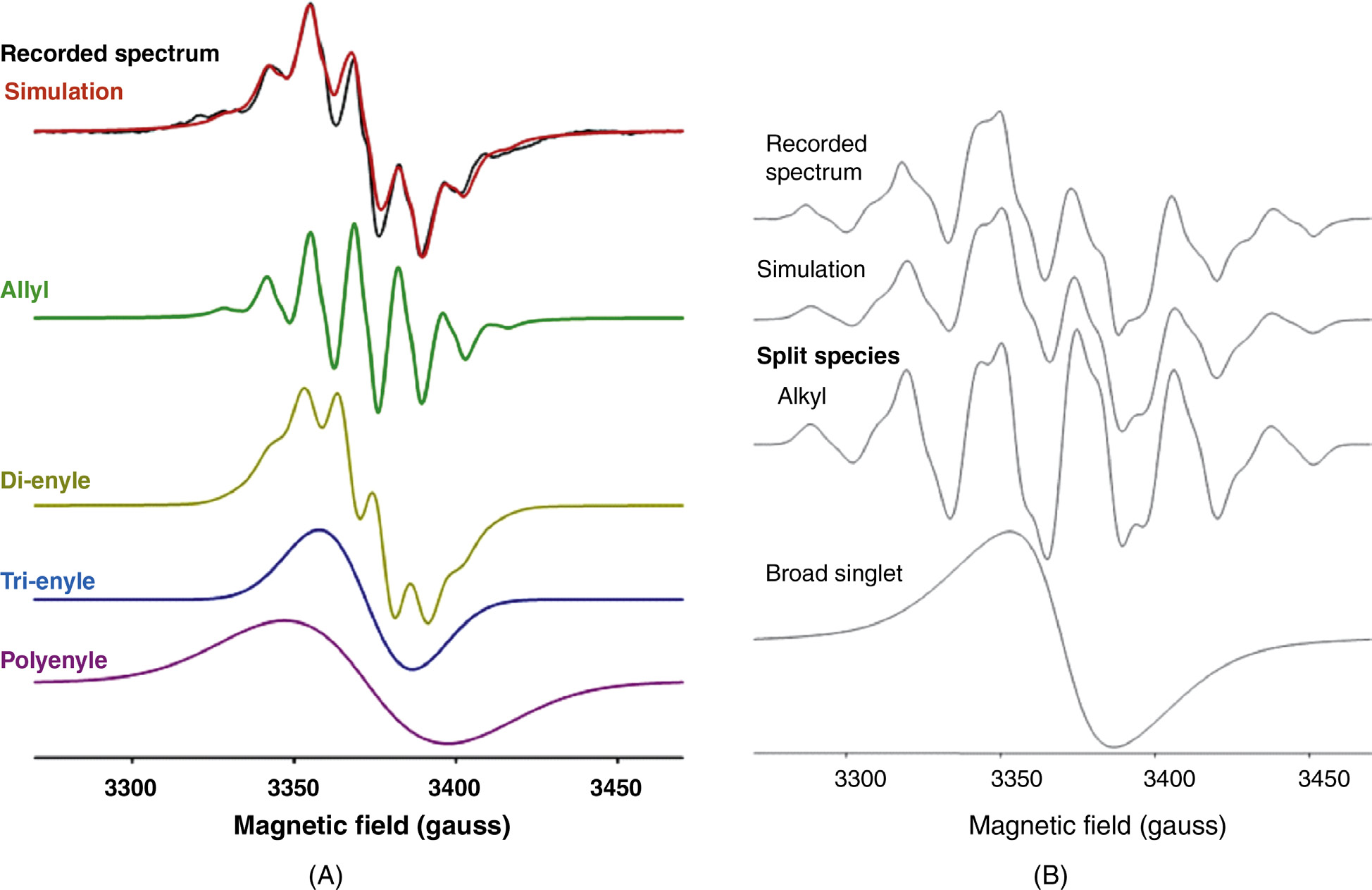
使用WinSim‐2002(Bruker)模拟程序,我们发现了烯丙基(n = 1)、二烯基(n = 2)、三烯基(n = 3)以及更高阶的多烯基(n > 3)自由基的浓度。主要由烯丙基贡献的电子自旋共振谱模拟如图35.5A所示,而主要由烷基贡献的模拟谱如图35.5B所示。表35.1列出了这些分裂自由基的丰度和超精细耦合常数。应注意的是,烯丙基谱的模拟可能未包含烷基,而烷基谱的模拟也可能未包含烯丙基。
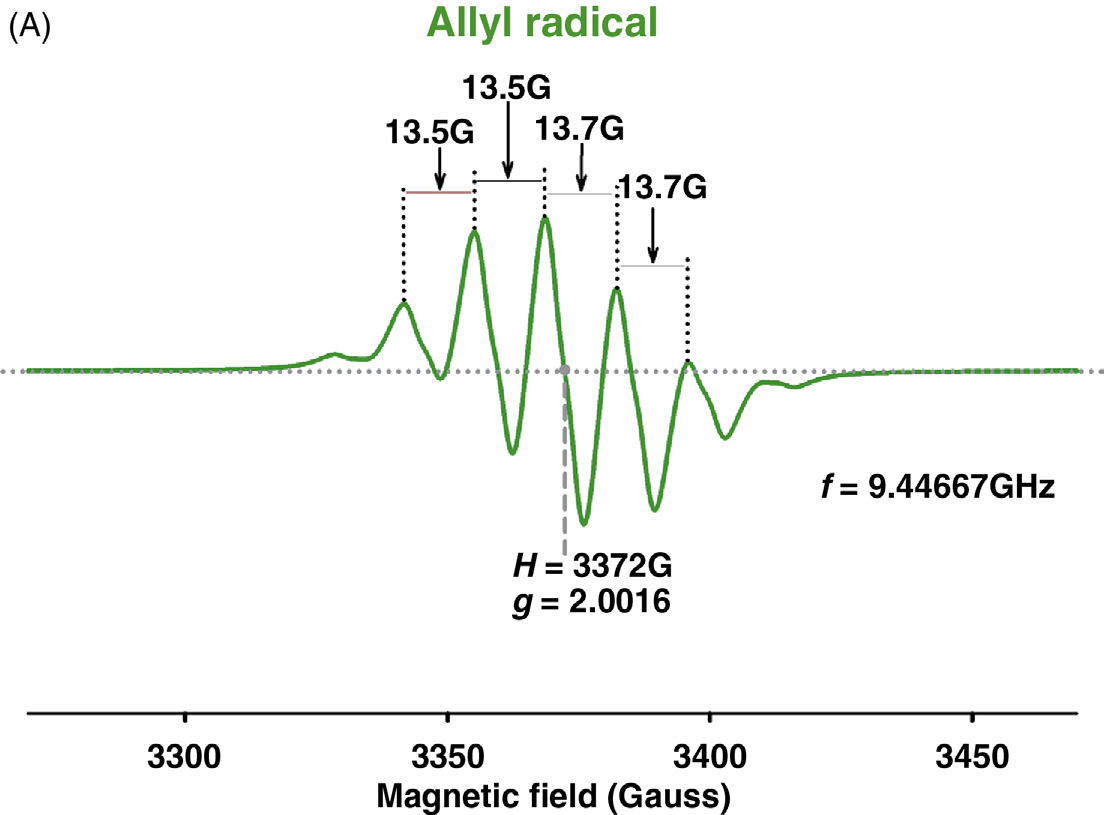
表35.1 模拟1和2中用于光谱模拟的丰度、自旋1⁄2超精细耦合常数、洛伦兹百分比和线宽
| 模拟1 | 模拟1 | 模拟1 | 模拟1 | 模拟2 | 模拟2 | |
|---|---|---|---|---|---|---|
| 烯丙基 | 二烯基 | 三烯基 | 单重态 | 烷基 | 单重态 | |
| 丰度 (%) | 23 | 17.70 | 26.70 | 32.60 | 25.70 | 74.30 |
| a-a | 2@13.3G | 3@8.9G | 4@7.7G | N/A | 1@22.1G | N/A |
| a-b1 | 1@4.0G | 2@1.9G | 3@0.7G | N/A | 4@31.6G | N/A |
| a-b2 | 4@13.3G | 4@8.9G | 4@7.7G | N/A | N/A | N/A |
| a‐各向异性 | N/A | N/A | N/A | N/A | 1@7.2G | N/A |
| %洛伦兹 | 87.4 | 100 | 88.3 | 29 | 0 | 59.2 |
| 线宽 (G) | 4.3 | 6 | 7.4 | 18.8 | 4.28 | 23.4 |
35.3.4 超高分子量聚乙烯中的过氧自由基
过氧自由基(‐H₂C‐HC‐O₂●‐CH₂‐)是超高分子量聚乙烯(UHMWPE)中氧化过程的前驱体。它会导致氢过氧化物和羰基物种(>C = O)的生成,包括酯、醛、醇等。这些在UHMWPE中的非自由基或中性副产物或终产物可通过傅里叶变换红外光谱技术(FTIR)进行检测或定量。实际应用中,通常利用羰基在1680–1720 cm⁻¹附近的傅里叶变换红外吸收带来测量UHMWPE中的氧化程度,并称之为氧化指数(OI)。电子自旋共振(ESR)是唯一能够直接检测固体或液体中过氧自由基的技术,但由于复杂聚合物基质中存在不同自由基的重叠信号,在应用于UHMWPE时这种检测具有挑战性。此外,由于测试样品从表面到核心的自由基在有氧或无氧条件下发生持续反应,每个单独的信号也随之发生变化。
基于实验数据,关于超高分子量聚乙烯中过氧自由基的讨论和推测已有报道,但据我们所知,尚未有明确的电子自旋共振证据被报道。许多研究者将所谓的单线谱归因于过氧[8,17–20]。然而,该单线谱也可能是由两个相互重叠的独立谱组成,每个谱均来源于非过氧自由基。在本节中,我们将介绍一种在本实验室最近成功检测超高分子量聚乙烯中过氧自由基的电子自旋共振方法。
35.3.4.1 超高分子量聚乙烯中过氧自由基的电子自旋共振证据
由于氧分子‐未成对电子(‐O₂●‐)相对于聚乙烯分子轴的独特取向,过氧自由基在电子自旋共振谱中产生(理论上的)不对称谱线,具有两个g值,g∥和g⊥。Ranby和J. F. 拉贝克讨论了电子自旋共振谱对聚四氟乙烯(PTFE)中过氧端基取向的依赖性[21]。在Rabek的综述中,给出了聚丙烯‐过氧自由基清晰的电子自旋共振谱,可视为过氧自由基的“指纹”(见[2], 第3章,第88页),但对于聚乙烯过氧自由基(低密度聚乙烯),其不对称谱线并不那么明显(见[2], 第3章,第74页)。为了更好地理解,建议读者参考J. F. 拉贝克汇编的关于聚烯烃(包括聚乙烯)中过氧自由基生成、链增长和终止机理的综述([2], 第3章,第67–102页)以及多尔([1], 第13章,第263–279页)。
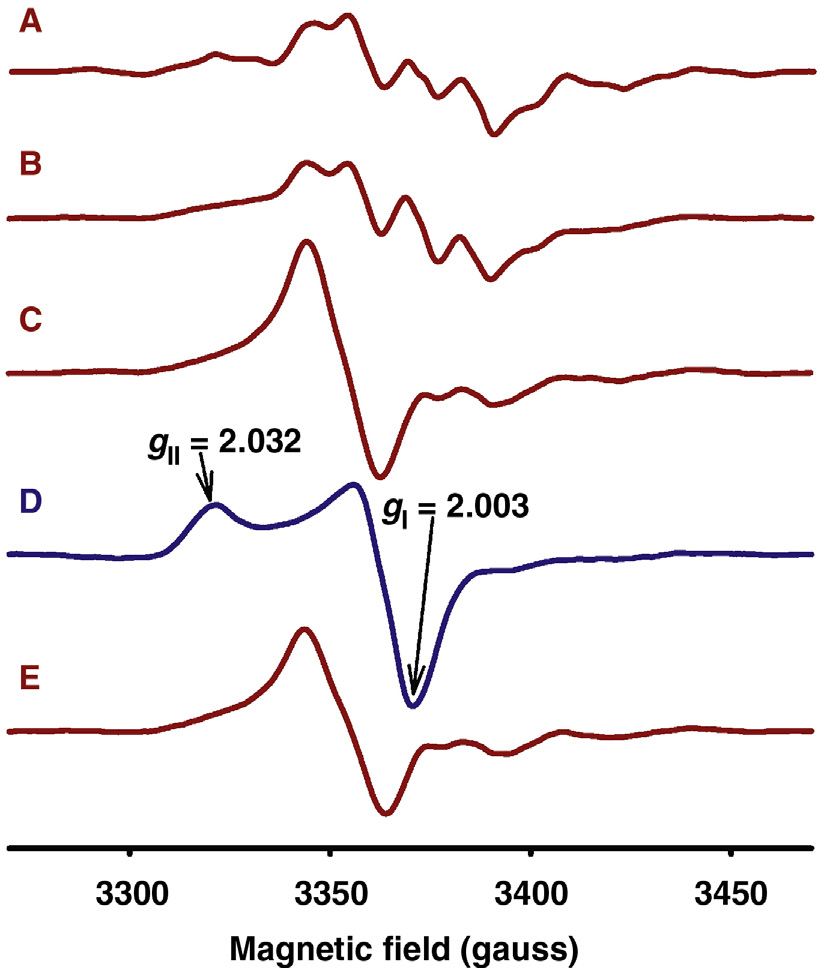
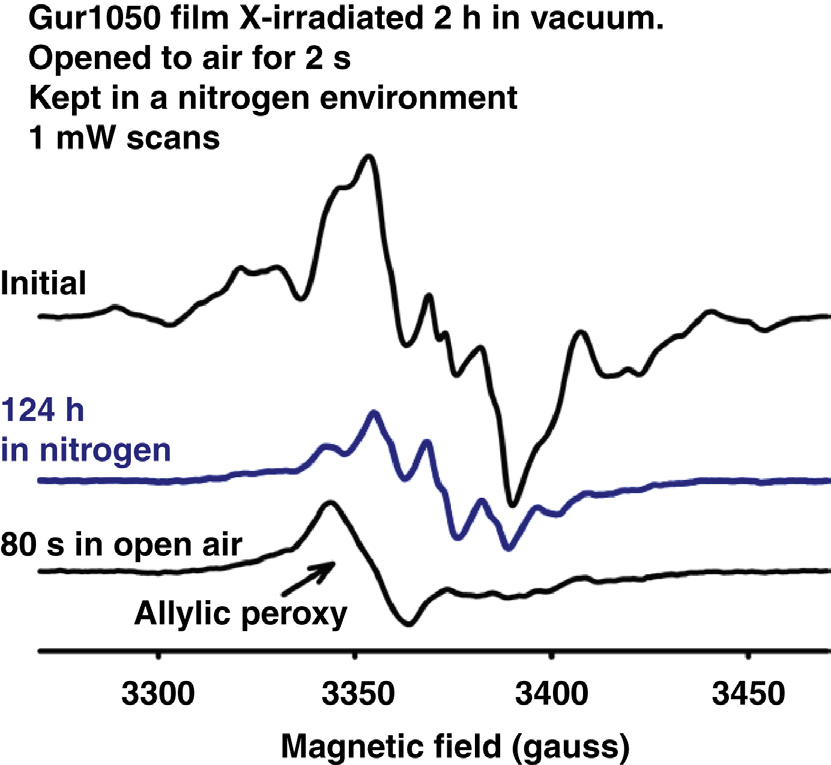
在我们的实验室中,于2004[22]首次直接检测到了UHMWPE中的初级过氧自由基。我们使用了200‐mm厚的样品(GUR 1050),并在氮气或真空条件下用X射线对其进行辐照。辐照后立即把样品转移到持续通入氮气的ESR腔中。结合微波功率饱和技术(PST),在室温及更低温度下记录电子自旋共振谱。在不同时间间隔,停止氮气流以使氧气(空气)与自由基发生反应。如图35.6所示,当样品暴露于空气1分钟后,将微波功率增加至32 mW(图35.6B),由烷基和烯丙基产生的共振线被抑制(饱和),并形成一条宽而不对称的信号(单线)(图35.6C)。尽管这并非过氧自由基的特征指纹,但它表明可能生成了过氧自由基。当通过向腔体内通入冷氮气将样品温度降至118 K(停止空气流动)时,获得了该特征指纹。电子自旋共振信号中明确的过氧结构由其清晰定义的g∥和g⊥位置(g∥ = 2.032和g⊥ = 2.003)所证实,如图35.6D所示。当温度升高至293 K(室温)并保持氮气流时,谱图(图35.6E)恢复为其原始结构(图35.6C),符合预期。这些测量结果表明,室温下出现的宽而不对称的单线可作为存在过氧自由基的指示剂;然而,仍需确认应通过在低温下记录电子自旋共振信号来进行。
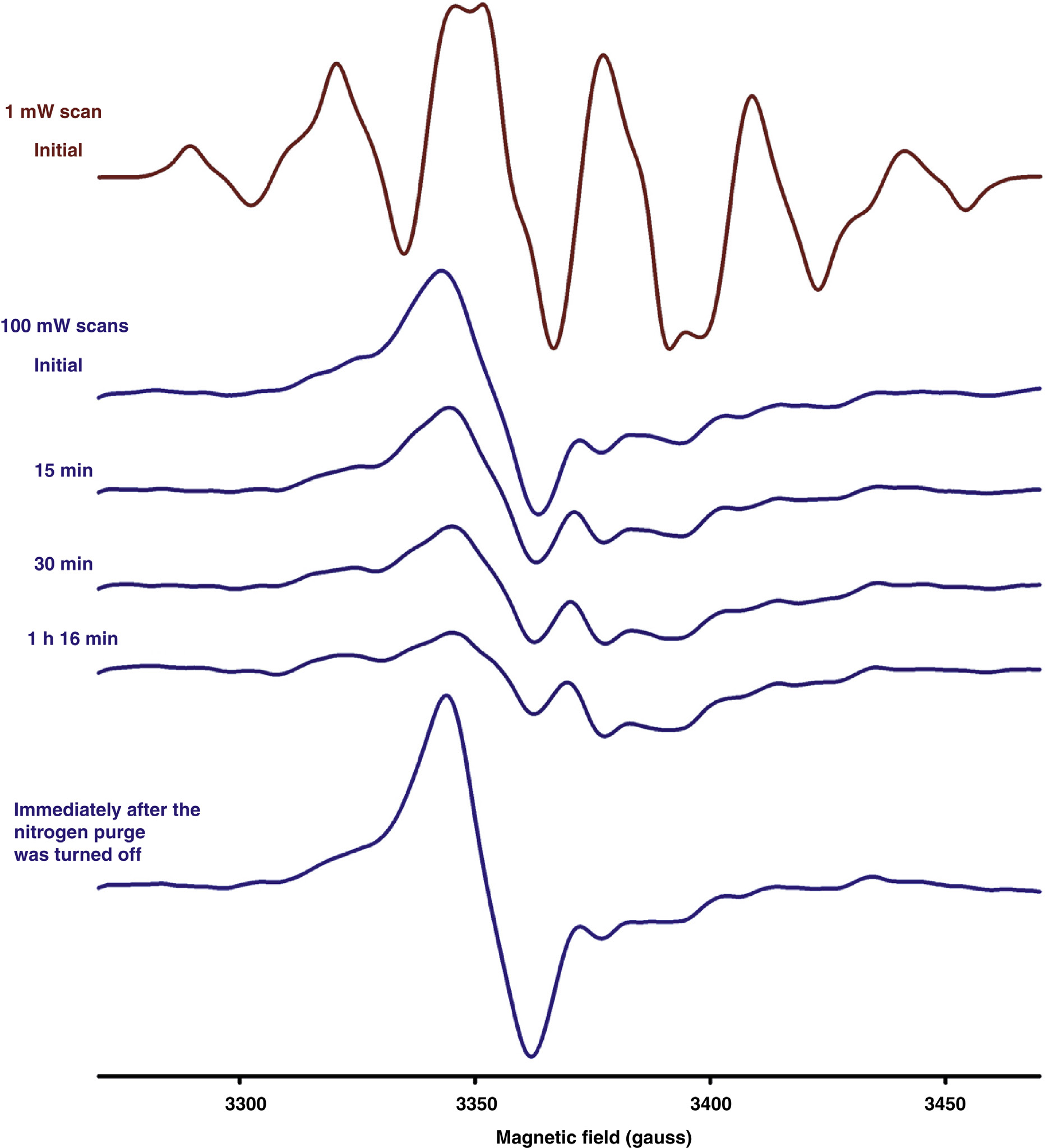
如图所示图35.7为在主要含有烷基自由基的样品上进行测试时获得的类似结果(1‐mW扫描)。在100 mW微波功率下,烷基自由基信号被饱和,过氧自由基信号显现出来。由在15分钟、30分钟和76分钟(1小时16分钟)记录的信号强度降低可以看出,在室温下的氮气环境中,过氧自由基浓度通过复合反应逐渐降低。当样品在氮气中放置76分钟后暴露于空气中时,过氧自由基信号强度增加,表明生成了新的过氧自由基。需要注意的是,烷基自由基在高微波功率下可能不产生电子自旋共振信号,但它可以与体系中存在的氧气发生反应并形成过氧自由基。
如图35.8所示,通过首先让烷基自由基在氮气中124小时内衰变为烯丙基,然后使其与氧气反应,证明了烯丙基生成过氧自由基的过程(空气)。相应的电子自旋共振谱如图35.8所示。
35.3.4.2 超高分子量聚乙烯中过氧自由基的半衰期
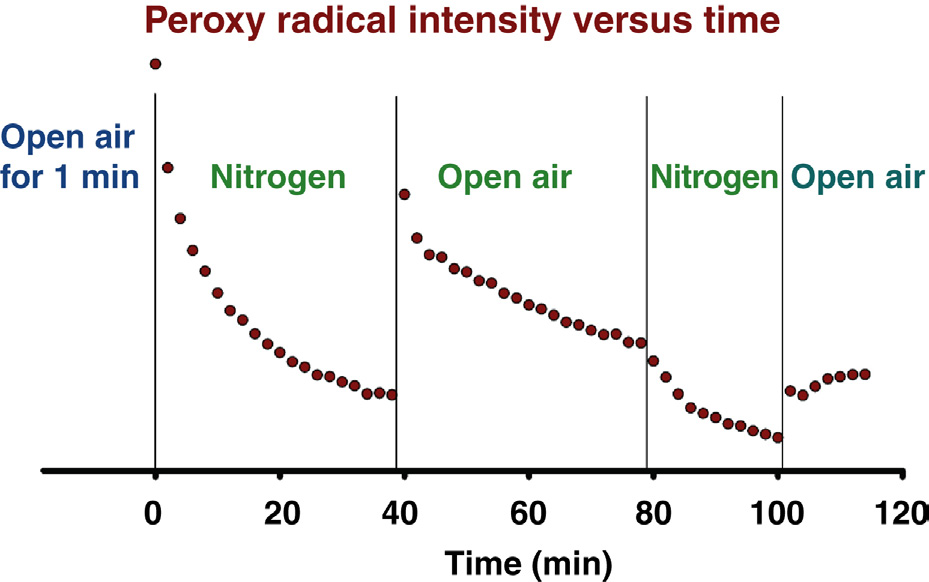
通过测量相应电子自旋共振谱的归一化峰‐峰高度,监测了过氧自由基在氮气中的衰变和在空气中的生成。
在氮气中的衰变比在空气中的衰变更显著,这可能是因为已存在的过氧自由基继续衰变,而在没有氧气的情况下几乎不会生成新的过氧自由基。然而,在开放空气中,新的过氧自由基可以生成,同时其他自由基发生衰变。当大部分过氧自由基在80–100分钟的时间间隔内衰变后,将同一试样再次暴露于氧气(空气)中,会导致产生少量新的过氧自由基,这由强度‐时间曲线的正向趋势表明。因此,超高分子量聚乙烯中的过氧自由基寿命非常短,半衰期为10–20分钟。根据奥尔巴赫和桑德斯[23]的研究,多尔得出结论:线性聚乙烯中过氧自由基在室温下的半衰期为20分钟,这一结果与我们在超高分子量聚乙烯中得到的结果非常接近[1](图35.9)。
35.4 超高分子量聚乙烯中的长寿自由基
35.4.1 长寿命初级自由基:其对温度和环境的依赖性
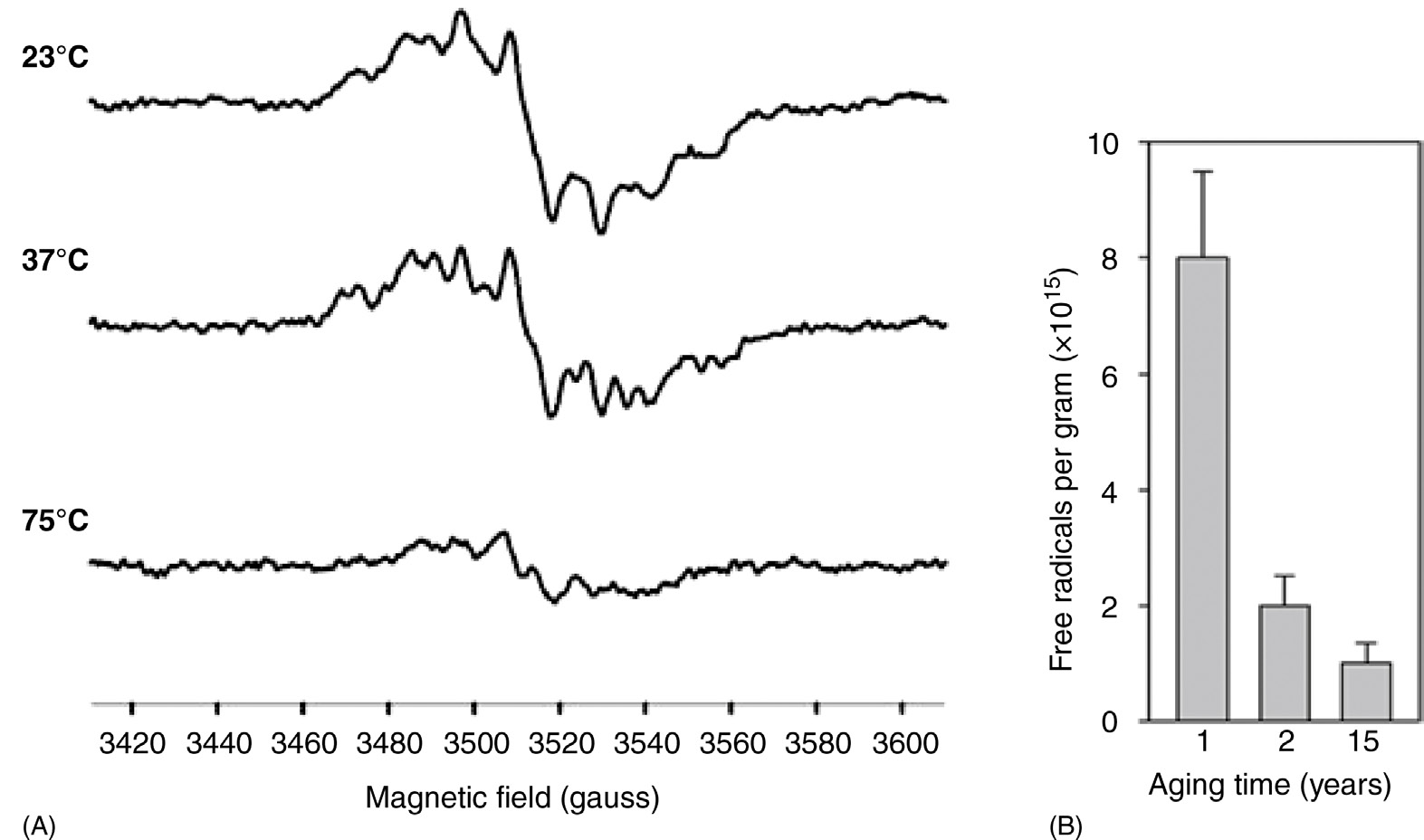
在惰性环境中辐照并储存的超高分子量聚乙烯(UHMWPE)中初级自由基的寿命如图35.10所示。UHMWPE样品被密封在电子自旋共振(ESR)样品管中,管内为真空或含有氮气或氩气。辐照(30千戈瑞伽马射线)后,样品管在不破坏密封的情况下,分别于体温(37°C)、室温(23°C)和75°C条件下储存。总储存时间约为15年。如图所示,所有在惰性环境中储存的样品中仍显著存在自由基——尽管其数量较15年前原始状态已减少了90%以上。在37°C储存的样品表现出与23°C储存样品相似的减少程度,而在75°C储存的样品则表现出最大的减少。总体而言,所有样品在惰性环境中储存的样品在电子自旋共振谱(图35.10A)中未显示出显著的结构变化,表明这些谱图确实来自初级自由基;但在浓度上均表现出整体下降(图35.10B),且在氮气、氩气或真空储存环境中,浓度下降程度没有显著差异。
所有在惰性环境中储存的样品中的自由基均为未与氧气反应的聚乙烯分子的自由基。由于自由基数量随时间减少,表明聚乙烯自由基之间发生了相互反应,这可能导致随时间推移进一步交联。此外,在低于75°C的惰性环境中对超高分子量聚乙烯进行以消除自由基为目的的热处理,并不能完全消除所有自由基。在温度低于75°C的惰性环境中存在的自由基。
35 电子自旋共振对超高分子量聚乙烯中大自由基的研究
35.4 超高分子量聚乙烯中的长寿自由基
35.4.2 长寿命氧化诱导自由基
除了短寿命的过氧自由基外(参见第35.3.4节),当初级自由基衰变时,辐照后的超高分子量聚乙烯的电子自旋共振谱中长寿命自由基的增长变得明显。由于这种长寿命自由基仅在氧气存在下,这种自由基被称为OIR(氧化诱导自由基),尽管这是在没有更好术语情况下的称呼。虽然我们已在大量样品中观察OIR超过20年,但其寿命仍未知。作为聚合物体系中的终端种类,OIR也被称为“终端”或“残余”自由基。然而,必须区分长寿命的OIR与在惰性环境中经长期辐照后老化后存在于UHMWPE中的“残余初级”自由基(>10年,在本实验室条件下)。仅有少量初级自由基能够在惰性环境中的长期老化过程中存留下来;老化时间越长,浓度越低(见第35.4.2.1节)。这些自由基也被标记为“残余”或“残余初级”自由基。
35.4.2.1 超高分子量聚乙烯中长寿命自由基的电子自旋共振证据
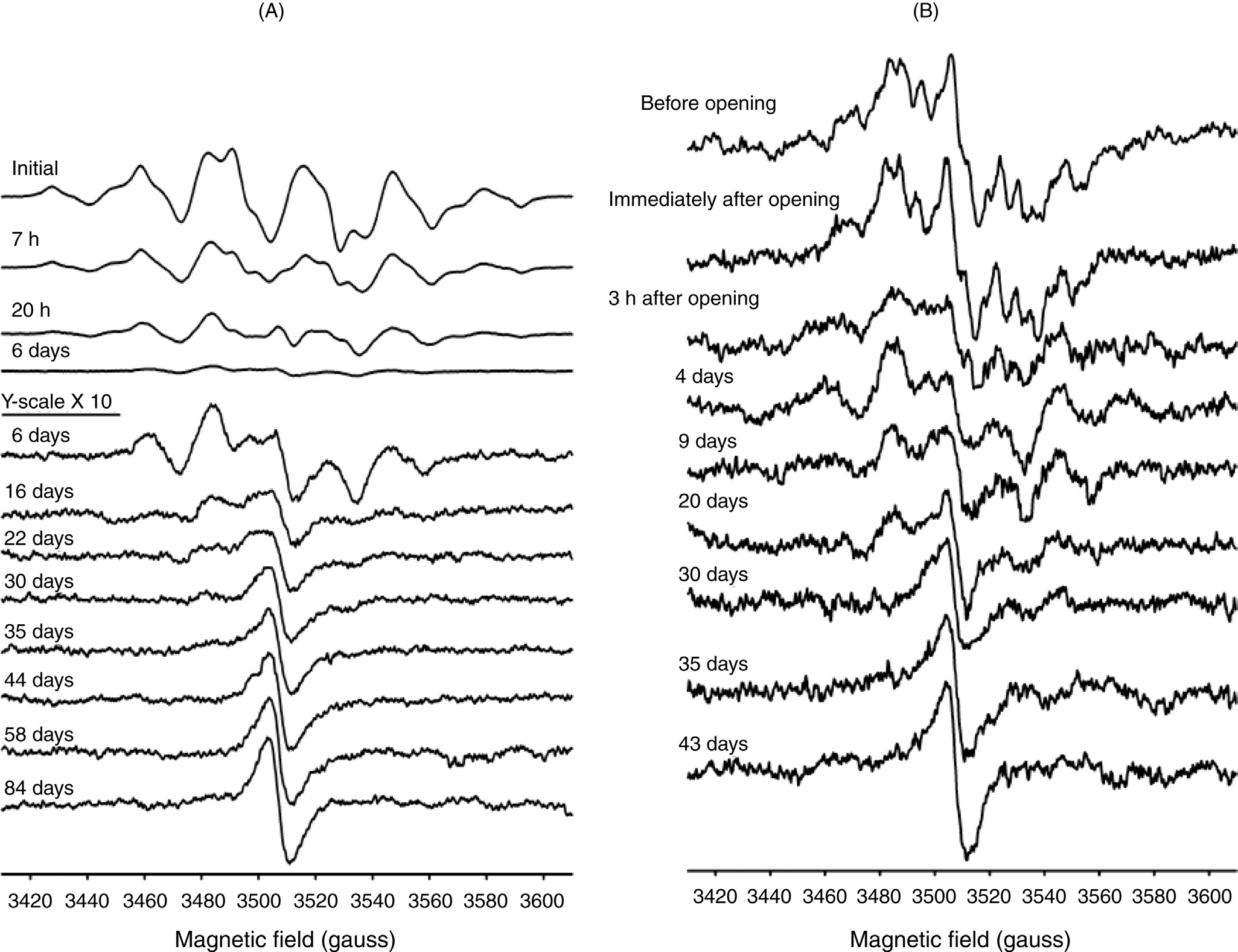
初级自由基在氧气存在下衰变后,OIR的增长情况如图35.11所示,其中在室温空气条件下,对两个不同样品A(GUR 4150)和B(GUR 4120)的ESR谱随时间的变化进行了记录。样品A在室温空气中用X射线辐照,并在辐照后立即开始进行ESR测试。注意,样品A的初始ESR谱类似于图35.2中所示的烷基自由基谱。样品B在室温真空中经γ辐照后,在真空条件下于75°C老化6年,随后暴露于空气中以发生氧化。样品B在暴露于空气前所记录的ESR谱非常弱,但显示出初级烷基/烯丙基自由基的特征(见图35.4A和B)。值得注意的是,样品B中的初级自由基在老化期间显著衰变,很可能通过自由基–自由基复合反应所致,这由微弱的ESR谱得到证实,但在老化期间并未转化为其他种类。在氧气存在下,这些长寿命的初级自由基(“残余初级”自由基)在样品B中发生衰变,同时OIR以与样品A相同的方式增长;OIR的增长时间约为35天。OIR的增长表现为ESR谱中约3500 G附近宽线的出现以及初始共振线的消失,如图35.11所示。根据这些以及通过对大量UHMWPE样品(包括GUR 4120、GUR 4150、GUR 1020、GUR 1050和Himont 1900)进行电子自旋共振测量所获得的类似结果,经X射线、伽马射线或电子束辐照,我们得出以下观察结果:
- 在氧气存在下,UHMWPE树脂经X射线、伽马射线或电子束辐照后可形成氧化诱导自由基。
- 辐照样品暴露于氧气(空气)约35天后,可通过电子自旋共振检测到氧化诱导自由基。
- 氧化诱导自由基的浓度与初始氧化时存在的初级自由基浓度成正比。
35.4.2.2 超高分子量聚乙烯中氧诱导自由基的增长与衰变
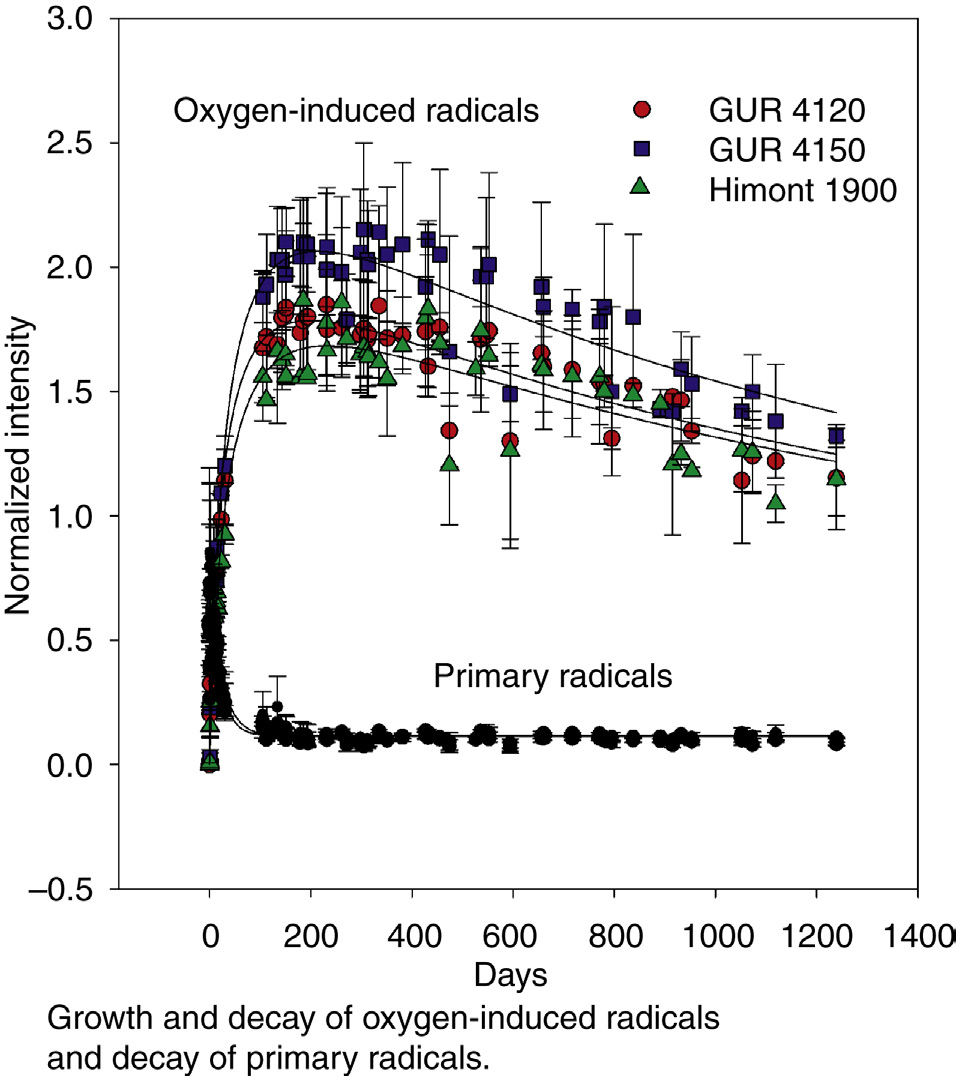
图35.12显示了三种样品GUR 4150、GUR 4120和Himont 1900在室温下经真空或氮气中γ辐照后暴露于空气中,其氧化诱导自由基(OIR)随时间的增长情况。该图中绘制了由OIR引起的电子自旋共振谱线的峰‐峰高度随时间的变化函数。如图35.12所示,OIR约在200天时达到最大值,随后缓慢衰减,因此具有非常长的寿命。目前尚不清楚为何GUR 4150的浓度(电子自旋共振谱线强度)始终高于GUR 4120或Himont 1900。
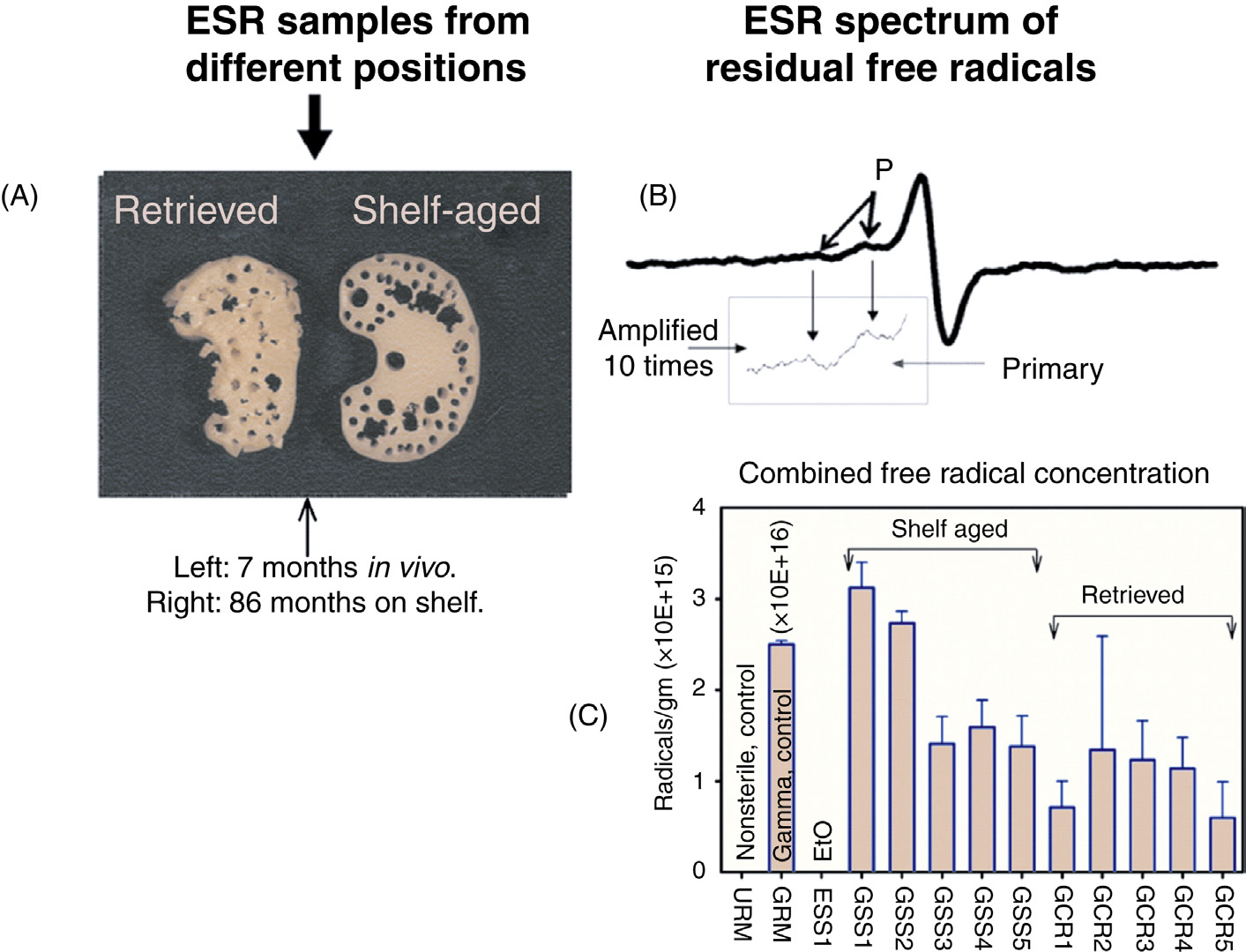
通过电子自旋共振可以证明在储存老化或回收的髋关节或膝关节部件中存在长寿命的氧化诱导自由基。图35.13显示了关于γ‐辐照胫骨平台衬垫(TPI)的数据。图35.13A展示了两个TPI(GUR 4120)的图像:左侧为临床回收样本(GCR),右侧为货架老化样本(GSS)。每个TPI上的孔洞是取样punched out的位置,编号用于识别(未显示)。图35.13B显示了来自位置5的临床回收样本(GCR5)的代表性电子自旋共振谱。如插图所示,主谱肩部的弱谱线表明存在微量初级自由基。图35.13C中的柱状图显示了自由基浓度(氧化诱导自由基)在TPI中随位置的分布情况。平均浓度和标准偏差(误差棒)是通过测量特定区域内的3–5个样品确定的。作为对比,本研究还包括了γ‐辐照对照组(GRM)、非辐照对照组(URM)以及环氧乙烷(EtO)灭菌的货架老化样本(ESS)TPI。ESS样本未检测到任何自由基,而GRM的浓度高出一个数量级。在GCR和GSS样本中,自由基均呈现出随机分布,尽管GCR样本的平均浓度略低于GSS样本。GCR样本中较低的自由基浓度可能是由于摩擦过程中温度升高导致自由基发生退火所致。TPI与髋臼杯关节面在运动过程中温度升高的现象由戴维森等人讨论过[24]。
在另一个例子中,从一个在体内使用了6–8年后的取出的髋臼杯中检测到了氧化诱导自由基in vivo。该髋臼杯(GUR 4150)在植入前经空气环境中γ射线灭菌(30 kGy, 60Co)。如图35.14A所示,样品取自髋臼杯摩擦面周边区域。图35.14B显示了氧化诱导自由基电子自旋共振谱线的峰‐峰高度所反映的自由基浓度随样品位置的变化情况,包括取出时以及取出后储存13年的结果。本研究得出两点结论:(1)在取出后于空气中老化13年期间,氧化诱导自由基浓度降低了约一个数量级;(2)摩擦面上自由基浓度的分布模式(可区分低磨损和高磨损区域)未发生变化。Jahan等人[5]报道,低共振曲线高度区域对应高磨损区域,高共振曲线高度对应低磨损区域。
35.4.2.3 通过电子自旋共振识别超高分子量聚乙烯中的氧诱导自由基
如图35.11和图35.13所示,由OIR引起的电子自旋共振谱是一条宽的单线,∆Hpp为9高斯,其g值大约在2.001到2.005之间。文献中有关OIR的报道有很多[8,17–20]。大多数报告认为它是过氧自由基(POO·),但也有一些认为是烷氧自由基(PO·)或多烯自由基(P·)。其中一些报道的研究是在低密度聚乙烯上进行的,且氧化反应为通过将测试样品暴露于低于室温的氧气中进行研究。
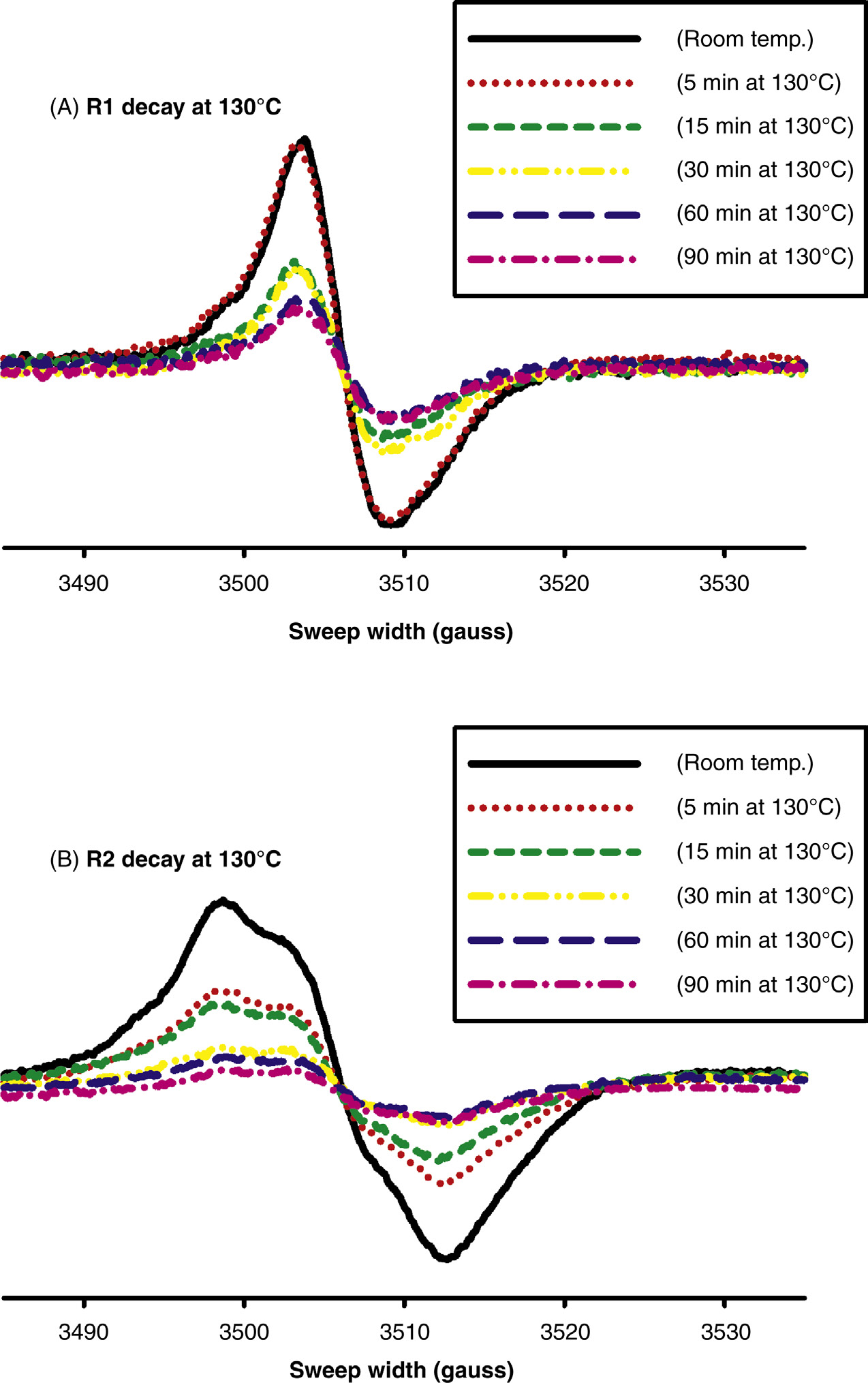
自2002年以来,我们的实验室(孟菲斯大学生物材料研究实验室)一直在探索确定UHMWPE中OIR结构的可能性[22,25,26]。使用高灵敏度的电子自旋共振波谱仪(布鲁克EMX300),在低温(118 K)下应用微波功率饱和技术,证明了该单峰由两种不同自由基种类R1和R2的重叠谱组成。通过在130–140°C进行热处理,并在极低(针对R1)或高(针对R2)微波功率下进一步区分这两种种类。本研究使用了高剂量(1000千戈瑞,钴‐60)样品以获得强烈的电子自旋共振信号。样品(GUR 4150)于1988年在空气环境中室温条件下辐照,而此项特定研究是在2004–2006年期间进行的。图35.15B显示了在标准操作条件(1.0毫瓦微波功率和1.0高斯调制幅度)下于室温(23摄氏度)检测到的典型单线电子自旋共振谱。该图进一步说明,在微波功率为0.01毫瓦时,室温信号变为明确定义的对称单峰(见图35.15A中的谱)。谱线的峰间距宽度∆Hpp = 5.5 G,且g值,g = 2.0044 ± 0.0003也已确定。该共振信号归属于一种碳中心自由基R1(见进一步讨论)。当微波功率增加至160毫瓦时,由R1引起的信号减弱,而谱图中(图35.15B)所示的弱肩峰增强(见图35.15C中的谱图)。该宽信号呈现出六条超精细线,在低温(−133°C)和10毫瓦微波功率条件下,R1的信号进一步减弱至无法检测的水平(见图35.15D中的谱图)。第二个信号(具有六个超精细谱线的一次导数谱)归属于第二种自由基R2。微波功率饱和测试表明,R2是氧中心自由基,而R1是碳中心自由基,因为前者在比后者更高的微波功率下才发生饱和(见图35.16)。关于氧中心自由基的电子自旋共振信号强度与微波功率依赖性的讨论见参考文献[10]。通过在75、100、130和140°C加热样品,进一步区分了这些自由基。图35.17A和B分别显示了在130°C下随时间变化记录的由自由基R1和R2产生的电子自旋共振谱。在此温度下,R2的损失(衰变)比R1更严重,当加热90分钟后,R2的信号降至谱仪检测限以下。在140°C时,R1和R2均在约18分钟内完全衰变。该结果表明,R1和R2可能都被困在结晶区中;R1完全位于结晶区内,而R2部分位于结晶区外,属于悬挂自由基。因此,R1最可能的种类是多烯基自由基(‐·CH-[CH=CH-]m ‐),这类自由基具有大量(m)共轭双键。此类自由基可产生单线电子自旋共振谱,由于未成对电子的离域作用,无明显的质子分裂。自由基2可能是类似的自由基,但它延伸出或悬挂在结晶区之外,因此可在略低于熔点温度下退火。由于R2产生的电子自旋共振信号表现出质子耦合(∼6条谱线,a_H = 4.8 G),当在较低温度(−133°C)下使离域冻结时,这种耦合更加明显,这表明R2的共轭键数量少于R1。这意味着R2可能是一个二烯基或三烯基(m = 2或3)自由基,而R1可能是一个多烯基(m > 3)自由基。表35.2列出了R1和R2的电子自旋共振参数。
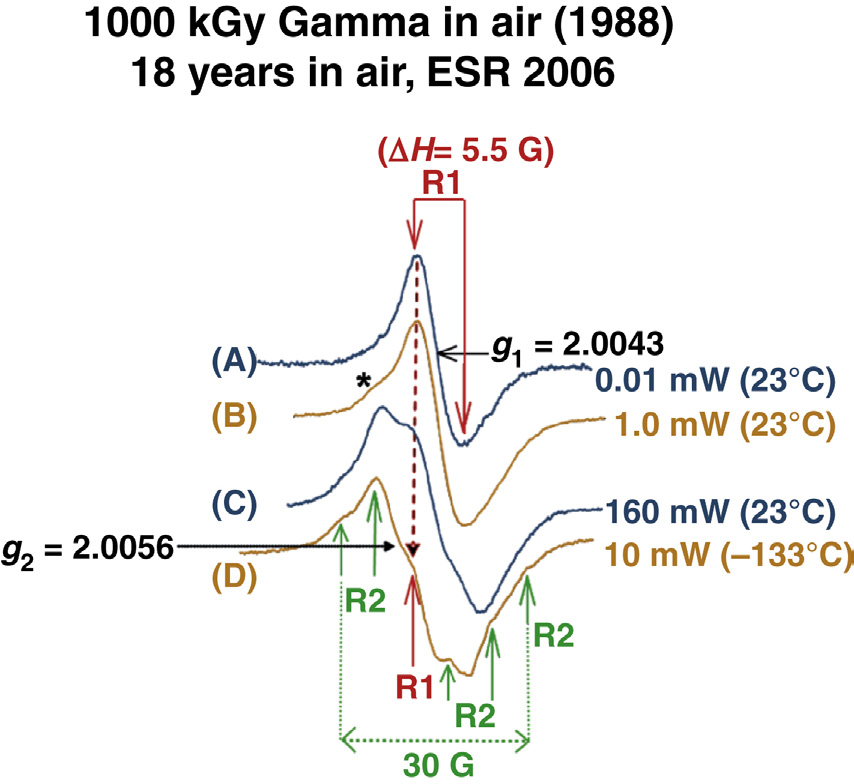
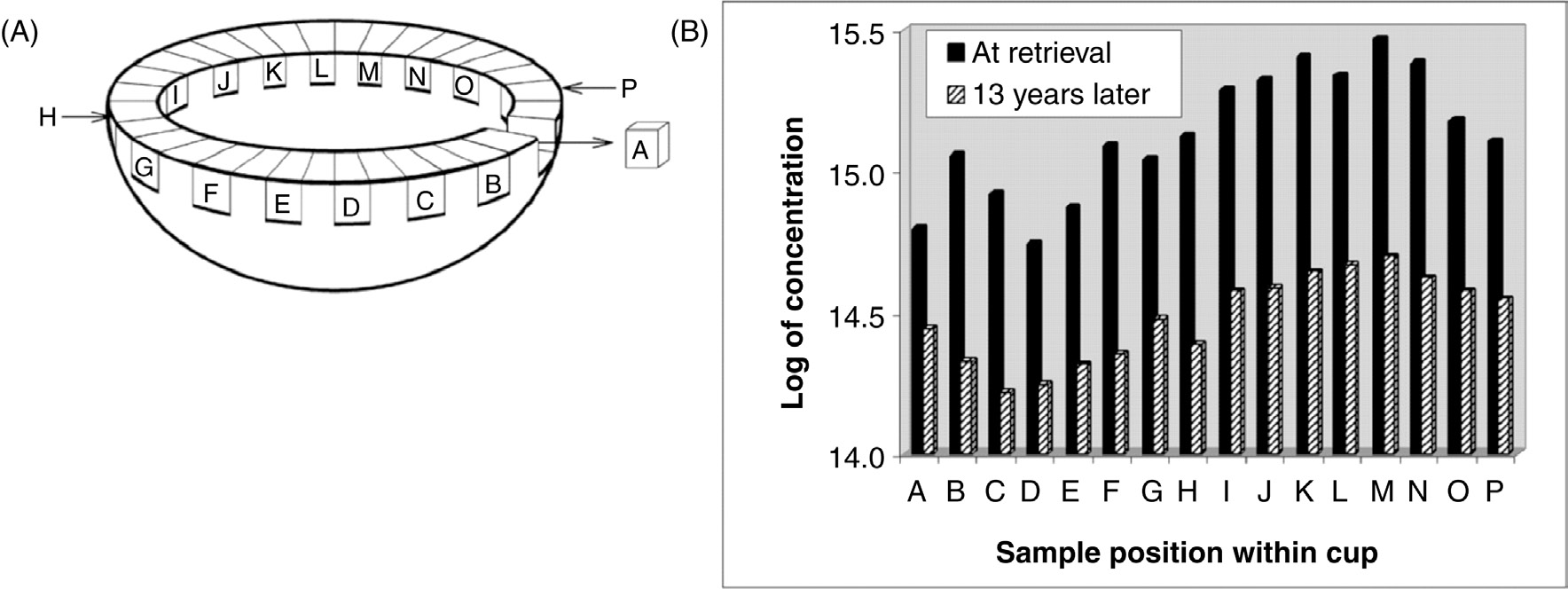
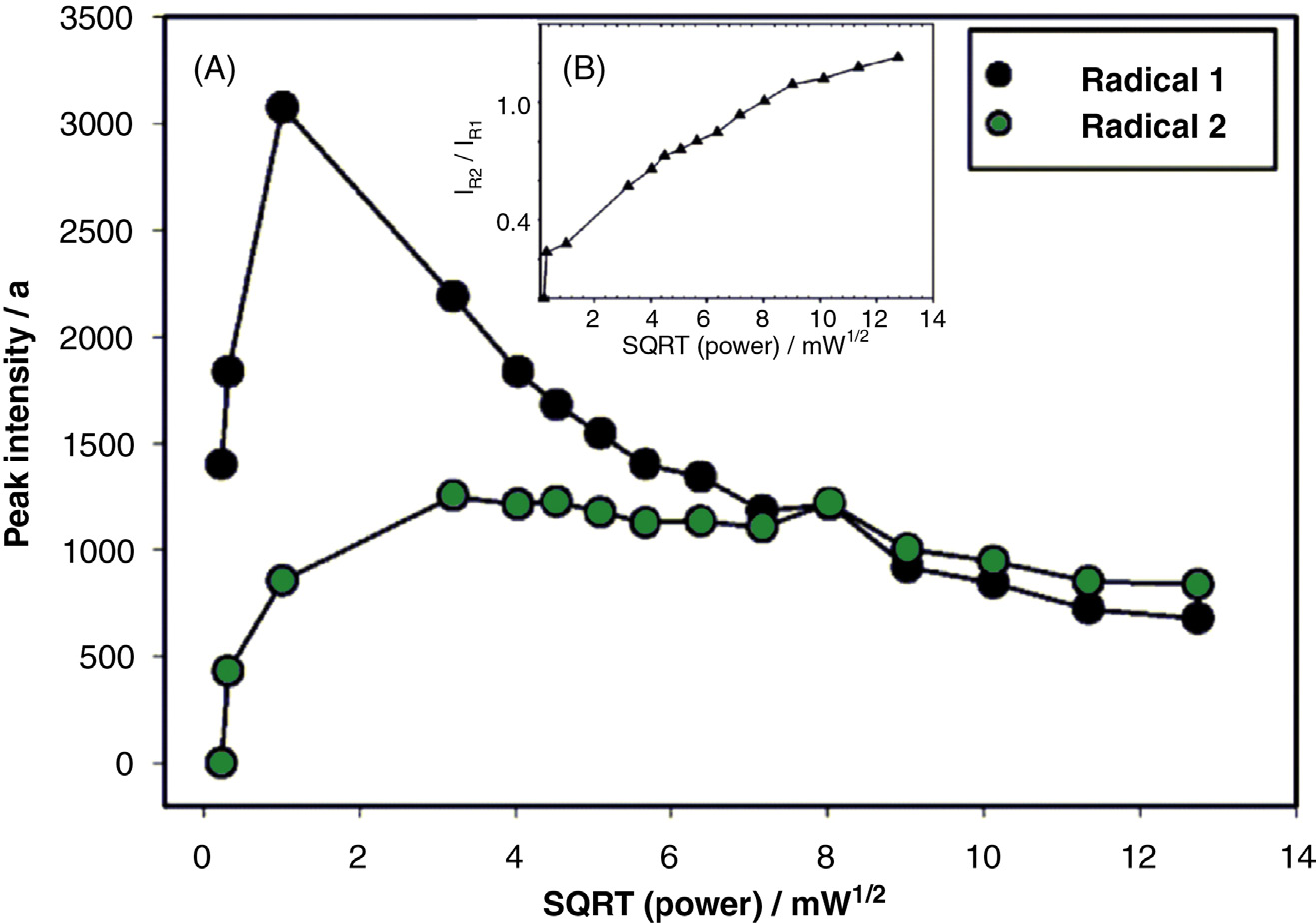
表35.2 自由基‐1(R1)和自由基‐2(R2)的电子自旋共振参数
| 自由基 | g值 | 谱线数 | 线宽 | 超精细常数,a_H |
|---|---|---|---|---|
| R1 | 2.0044 ± 0.0003 | 1 | 5.5 G | – |
| R2 | ∼ 2.0056 | 6 | 9.4 G | 4.8 G |
尽管这项研究的细节可以在文献中找到[22,25,26],但关于氧化诱导自由基的重要发现可以概括如下:
- 在氧气存在下,两种自由基(R1和R2)会长时间滞留在辐照的UHMWPE中(在本实验室中可达>20年)。
- 其中一种(R1)是多烯基P·(‐·CH‐[CH = CH‐]m ‐,其中m为> 3)。
- 第二种自由基R2是氧中心多烯基PO·(‐·OCH‐[CH = CH‐]m ‐,其中m为= 2或3)。
- R2是悬挂自由基;其所在区域的结晶区程度低于R1。
- 没有电子自旋共振证据表明存在长寿命的peroxy自由基。
35.5 超高分子量聚乙烯中的中间自由基
近年来,由于过氧基(POO●)和长寿命氧化诱导自由基可能产生的破坏性影响(氧化降解),超高分子量聚乙烯的电子自旋共振研究主要局限于对这两种自由基的研究。因此,中间自由基过程在很大程度上仍未得到充分探索。当然,文献[1,2,27,28]中可以找到描述聚乙烯自由基生成、链增长和终止过程的化学模型或反应机理。本节介绍辐照后超高分子量聚乙烯中某些中间自由基反应的电子自旋共振证据。
如图35.11所示,初级自由基在空气中衰变时,OIR的生成需要30–45天。有趣的是,经75°C加热6年后的真空密封样品中残留的“初级残余”自由基,在暴露于空气中后,也在相同时间内产生OIR。然而,后一种情况下的信号强度非常弱,这是因为在重新接触空气时初级RC较低。与氧气(空气)发生反应。这种情况表明,在真空或惰性环境中于75°C温度下加热,可以降低但无法完全消除辐照超高分子量聚乙烯组分的氧化潜能。在近熔点温度下加热或退火可能消除所有自由基,或将其浓度降低至电子自旋共振检测限以下。另一个重要观察结果是构成末端OIR(前文已讨论)的多烯基自由基并非初级或多烯基自由基,而是在氧气存在下由初级自由基反应生成的。
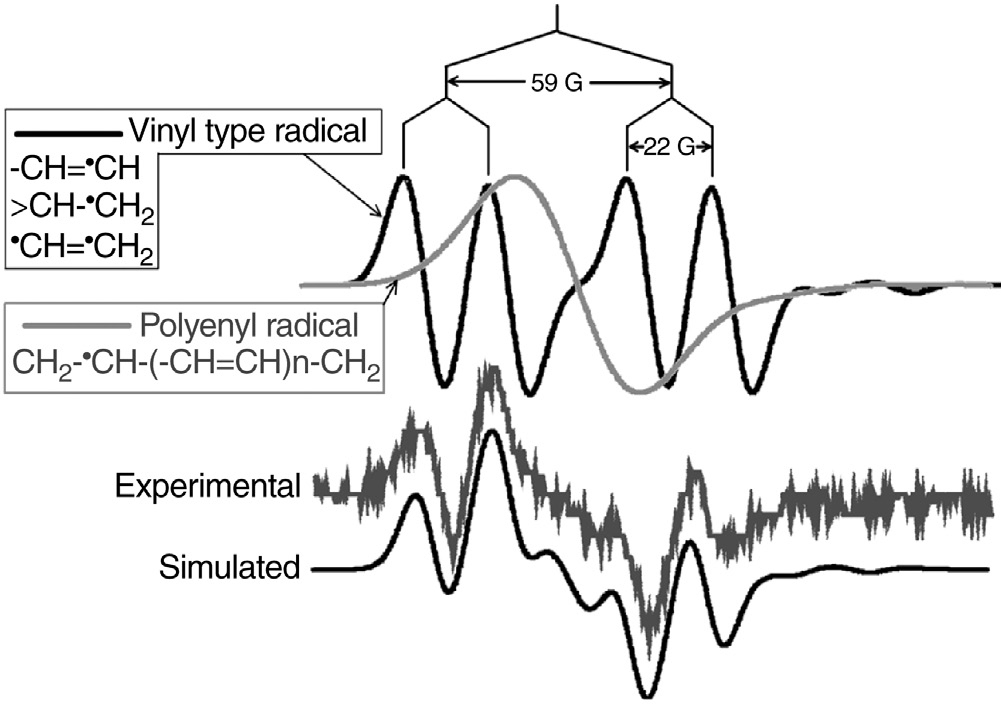
在最初的30–45天内,初级自由基在氧气存在下发生衰变,电子自旋共振谱随时间变化并变得非常复杂。电子自旋共振谱的复杂性源于以下事实:表面或近表面处的自由基(OIR多烯基和/或过氧基)、内部的自由基(初级烷基/烯丙基)以及介于表面与内部之间的其他未知的二级或三级自由基均在同一共振磁场区域内产生各自独立的谱。因此,对自由基类型进行明确鉴定变得非常困难。在我们的一项研究中,我们记录到一个电子自旋共振信号,并将其归因于乙烯型自由基。谱图中的乙烯特征在OIR信号出现之前变得清晰,此时烷基/烯丙基产生的信号已衰变或消失。为验证这一点,我们对该谱图进行了计算机模拟(见图35.18)。
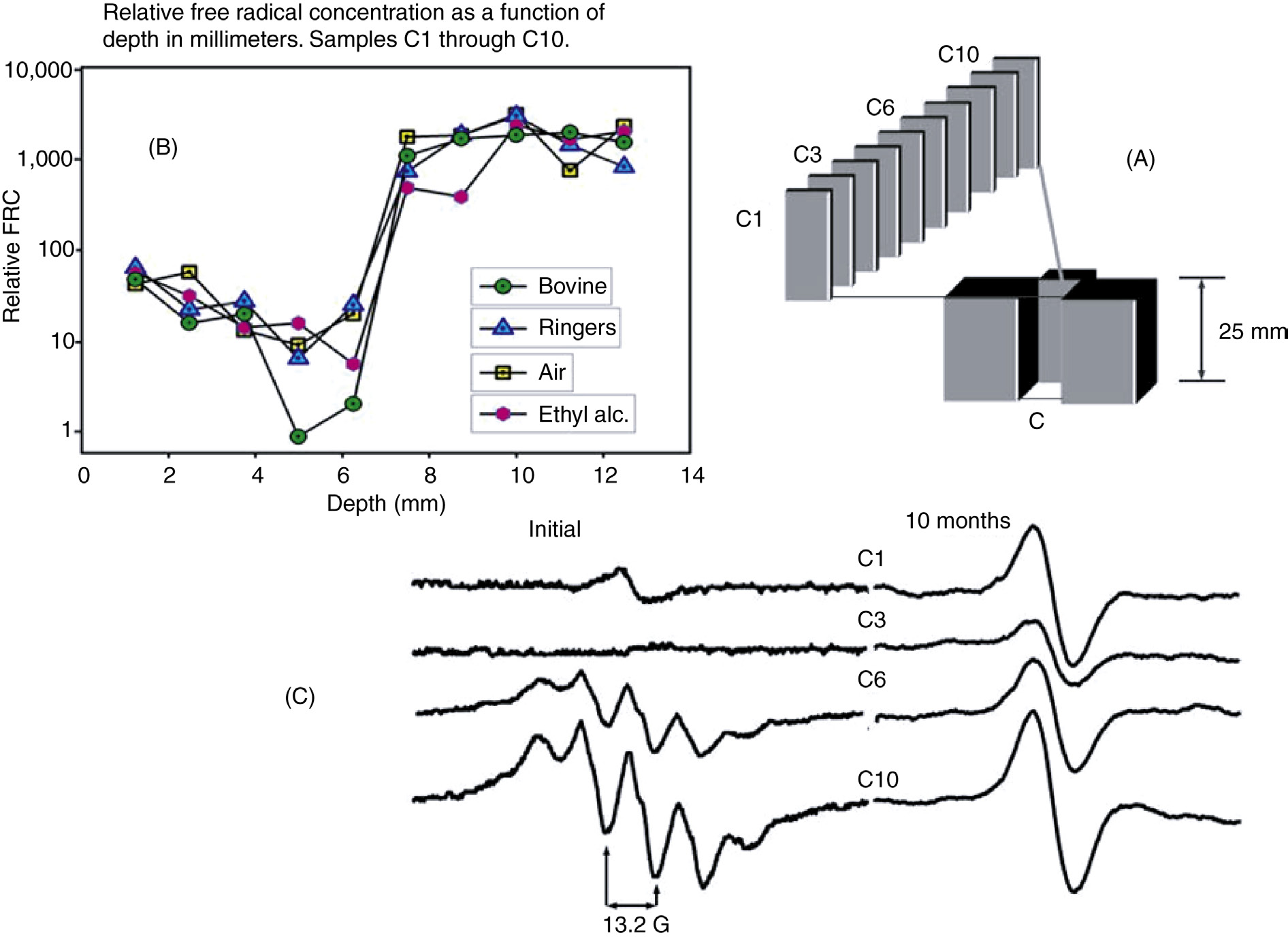
在另一项研究中,我们测量了一系列样品中的自由基浓度(RC),这些样品(1.20 ± 0.05 mm厚)取自一个1英寸立方体的超高分子量聚乙烯(GUR 4150)从表面到核心的不同位置。该立方体在室温下经空气环境中γ辐照(30 kGy, 60Co)后,分别在林格氏液、牛血清、乙醇或空气中老化1年。每个表面样品均显示出表面自由基(OIR)的存在,而核心样品则显示出初级自由基的存在。其余样品中也检测到了不同自由基的组合。在特定测量时间下,约5毫米深度处的自由基浓度最小或不存在(见图35.19)。测量后,样品在室温空气中存放10个月后再次进行测量。正如我们所预期的,每个样品均显示出相同的氧诱导自由基。图35.19还展示了典型的电子自旋共振谱。该研究得出结论:在辐照超高分子量聚乙烯固体中,初级自由基衰变和氧诱导自由基增长期间,表面与内部之间可能存在一个非自由基区域。进一步推测,非自由基区域可能是氢过氧化物种类所在的区域,而该区域无法通过电子顺磁共振检测到[29]。
35.6 维生素E掺杂的超高分子量聚乙烯
35.6.1 维生素E掺杂的超高分子量聚乙烯的电子自旋共振
维生素E(a‐生育酚,a‐T)已被发现可通过清除自由基来提高超高分子量聚乙烯的抗氧化性[30–32]。然而,关于a‐T抗氧化行为的大多数报告基于傅里叶变换红外光谱数据或力学测试结果,据我们所知,电子自旋共振数据极为有限。尽管本节提供了近期电子自旋共振研究的简要总结,但详细内容可参见里德利和贾汉的出版物[33,34]。有关抗氧化剂(生育酚或Irganox)的电子自旋共振的更多信息可从陈等人关于维生素E与脂质反应的报告中获得[35]和Walker等人[36],以及Jawor-ska等人在低密度聚乙烯上的研究[37],和山崎和濑口[38]。
如前所述,我们报道了a‐T掺杂的超高分子量聚乙烯粉末树脂(标记为“a‐T‐树脂”)和压制成型固体[33]的电子自旋共振结果。在实际应用中,为避免塑化作用,通常将a‐T的含量控制在1.0 wt%以下;然而,为了能够在初级或次级聚乙烯自由基的强共振信号存在下检测到a‐T自由基,我们使用了高达25% v/v的浓度。Jaworska等人[37]在低密度聚乙烯中使用了10% Irganox(一种抗氧化剂),以在液氮温度(77 K)下检测抗氧化剂自由基。
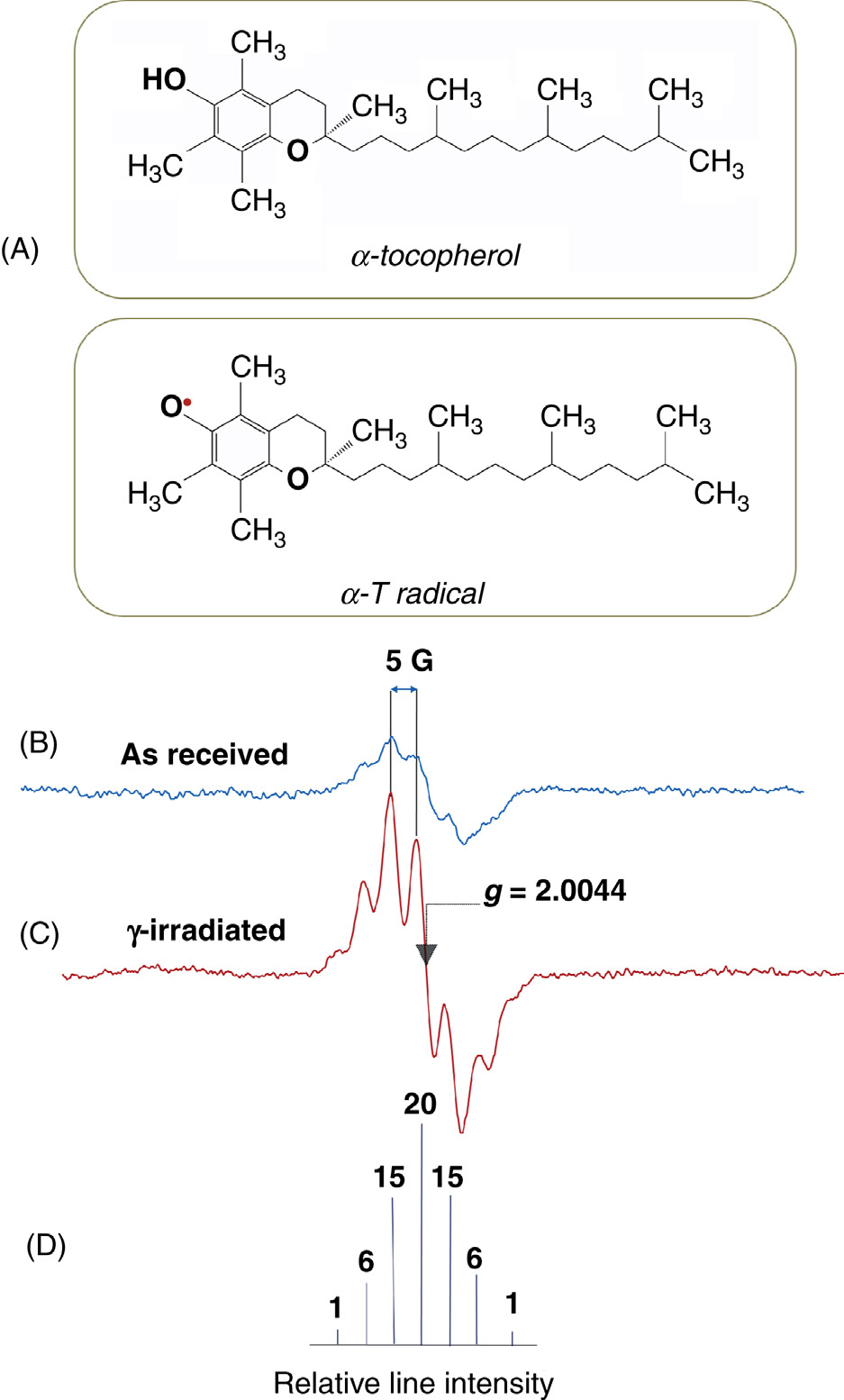
维生素E在室光或电离辐射下会产生自身的自由基(生育酚氧自由基,a-T-O.)。维生素E的分子结构如图35.20A所示,其自由基的结构如图35.20B所示。相应的电子自旋共振谱如图35.20C所示,显示了a-T-O.的电子自旋共振特征。图35.20C还显示由于a-T-O.自由基在as received维生素E中产生的谱。如图35.20C所示,一次导数ESR谱包含七条共振线,谱线间距为∆H = 5 G,线宽为∆W = 2 G,强度比为1:6:15:20:15:6:1(由图35.20D中的棒状图表示)。该自由基的特征谱分裂因子g值为g = 2.0044(由箭头指示)。这些数据与文献报道一致[35,40,41]。
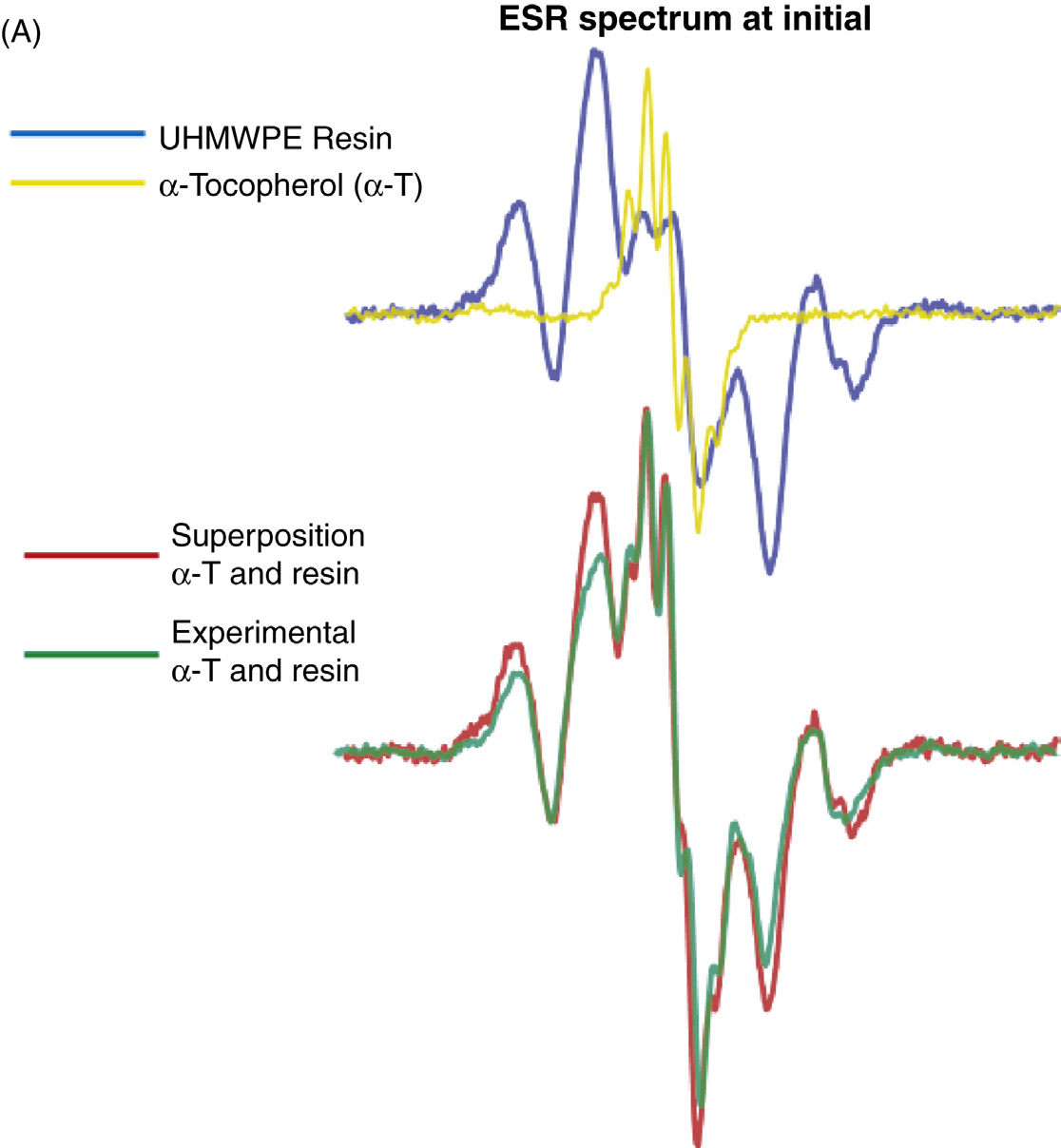
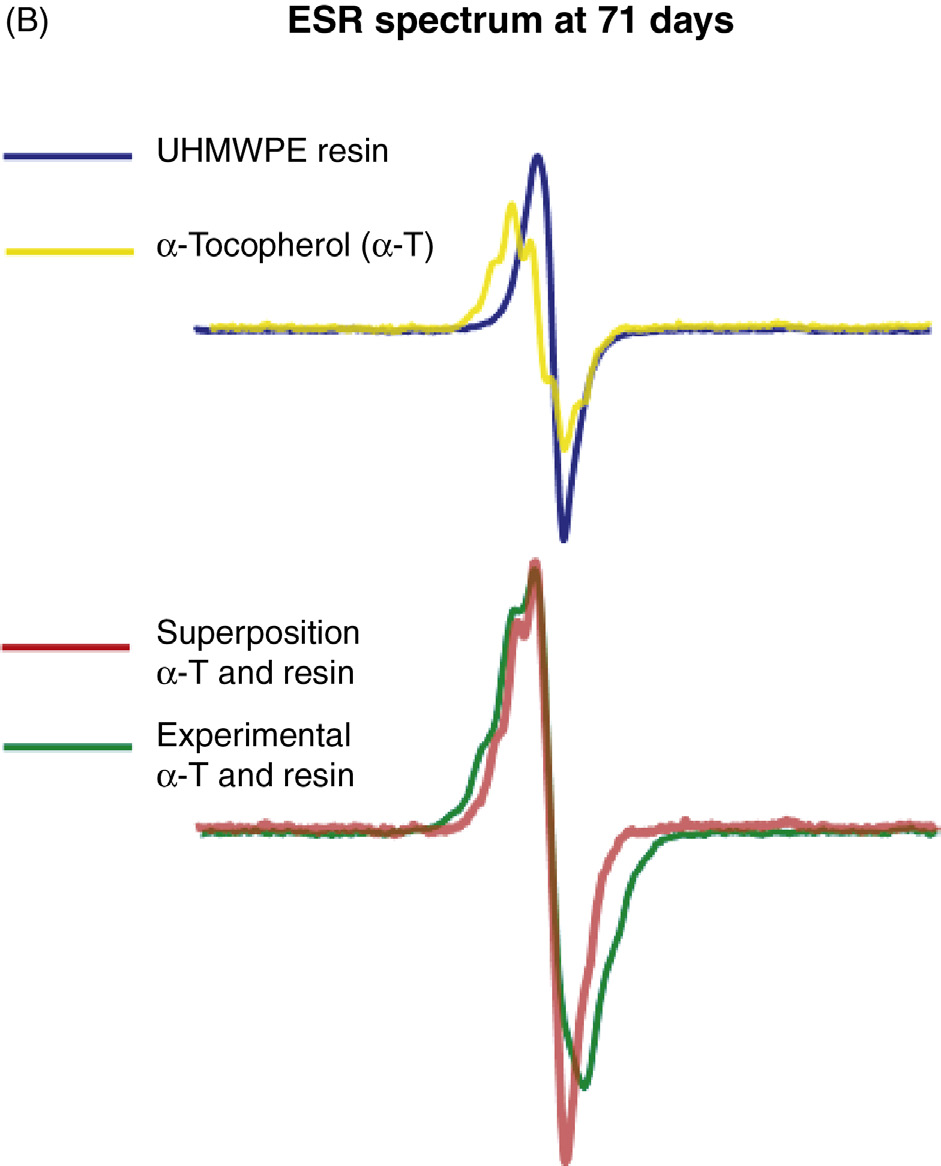
根据我们最近的报告[33],维生素E(a‐生育酚)在空气环境中对粉末样品(GUR 1050)进行γ辐照(30 kGy, 60Co)时,能够淬灭初级聚乙烯自由基,但在氮气环境中则未能淬灭任何自由基。当将辐照后的粉末从氮气中取出并置于空气中时,发生了自由基反应,并通过电子自旋共振检测到了氧化诱导自由基的形成。至于自由基在氧气(空气)存在下,含有维生素E的粉末样品与不含维生素E的粉末样品之间未发现可检测的差异。实验测得的电子自旋共振谱与计算机生成的电子自旋共振谱之间的良好拟合,如图35.21A所示(初始状态)以及图35.21B所示(71天后的最终测量结果),证实了维生素E(a-T-O.)自由基和树脂(初级或氧化诱导自由基)自由基是独立衰变的。本研究得出了三个观察结果。
- 就辐照后在空气中测得的电子自旋共振而言,氮气包装和开放空气条件下的a‐T‐树脂样品之间未发现可检测到的差异。
- 高浓度的a‐T使得维生素E自由基(a-T-O.的电子自旋共振信号)能够在聚乙烯自由基产生的相对较强烈的信号存在下被监测。这对于比较自由基‐自由基反应[37]具有潜在用途。
- 尽管超高分子量聚乙烯树脂粉末没有临床相关性,但粉末样品因其具有较大的有效表面积,使氧气(空气)易于接触,从而为研究氧气存在下的自由基反应提供了更好的手段。
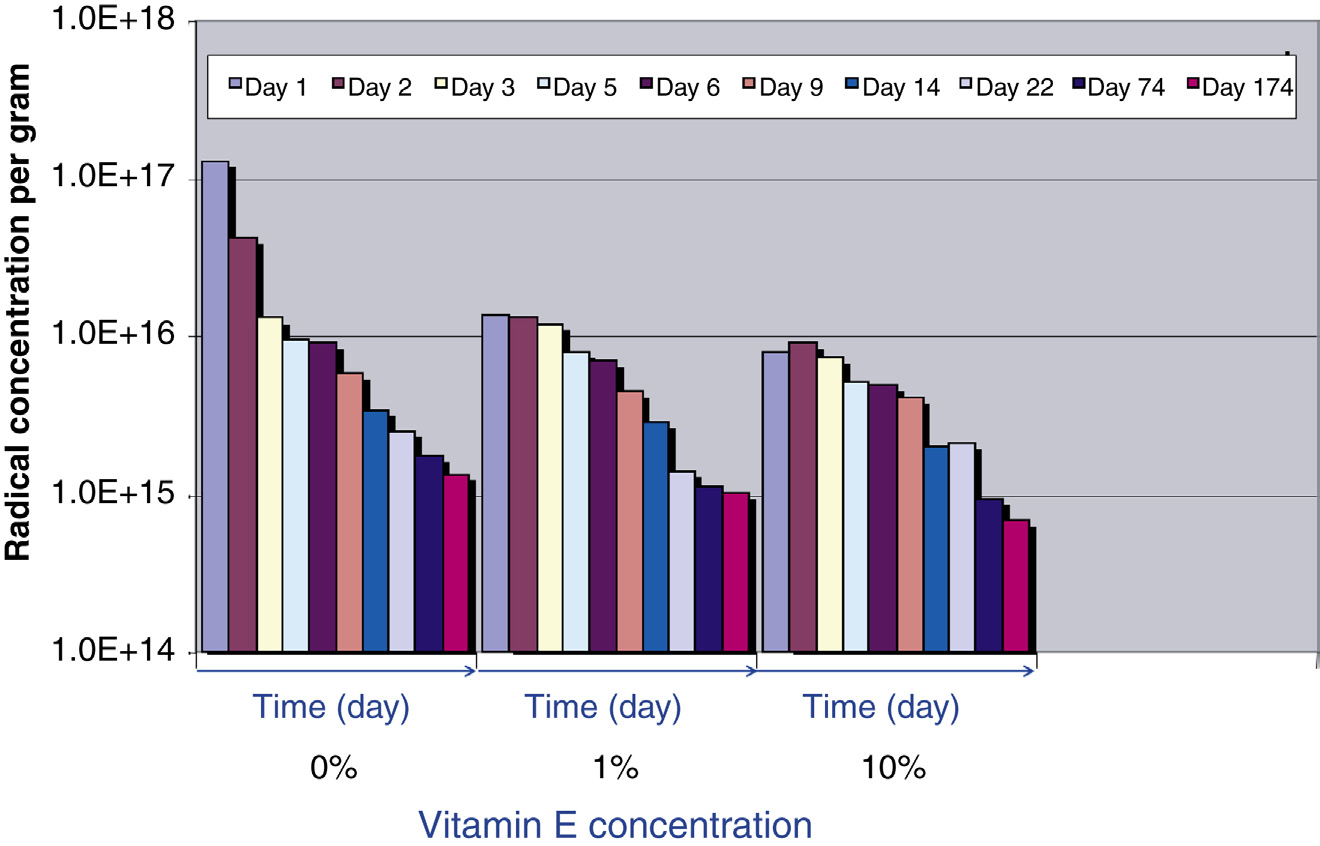
在含有0.0%(对照组)、1.0%或10.0%a‐T的压缩成型固体(UHMWPE,GUR 1050)中也观察到类似的辐照后氧化行为。在后续的衰减模式方面,对照组(0%a‐T)与含维生素E的UHMWPE之间未发现可检测的差异。然而,本研究中有一个重要发现:与对照组(0%a‐T)相比,所有UHMWPE‐a‐T样本中的初级自由基含量均减少了约一个数量级(见图35.22)。
由于缺乏初始数据点,推测a‐T对自由基的淬灭必须在辐照期间或辐照后立即发生。从供应商处接收辐照样本所需的时间(3‐5天)是辐照与电子自旋共振测量之间延迟的主要原因。需要进一步研究来解决此问题。
35.6.2 电子自旋共振法检测辐照维生素E-超高分子量聚乙烯共混物中的维生素E自由基
对于观察含有维生素E和UHMWPE自由基的电子自旋共振谱(如辐照后的维生素E/UHMWPE共混物),可通过调节电子自旋共振的运行参数,以更好地区分两者(维生素E自由基与UHMWPE自由基)。这主要通过调整微波功率和调制幅度来实现。例如,UHMWPE的主要烷基和烯丙基自由基的电子自旋共振信号强度随微波功率增加而减弱,而维生素E自由基信号则随微波功率增加而增强。图35.23展示了X射线辐照并在空气中放置约1小时后的维生素E共混超高分子量聚乙烯的三个谱图。注意当调制幅度从5 G降低到1 G(同时整体信号强度减弱)。当微波功率增加时,维生素E自由基谱会更加明显,从而相对于维生素E自由基(a‐TO*电子自旋共振谱线)的谱线变得更加清晰,而代表超高分子量聚乙烯的部分(用谱左侧的箭头和虚线表示)则相对减弱。
35.6.3 维生素E中储存辐照超高分子量聚乙烯的影响
我们研究了辐照超高分子量聚乙烯(UHMWPE)中的自由基活性,结果表明当UHMWPE与维生素E(α‐生育酚)接触时,反应速度减缓。将经30千戈瑞伽马射线辐照的UHMWPE分别储存在维生素E中,并与储存在氮气(无氧气)和空气中(含氧气)的样品进行比较。通过电子自旋共振(ESR)监测自由基活性,包括氧诱导自由基(OIRs)的形成,后者表明氧气与辐照过程中产生的初级自由基发生了反应。
如图35.24所示,保存在氮气中的样本与保存在维生素E中的样本结构非常相似,但保存在维生素E中的样本下降程度较小。这表明,保存在维生素E中的样本正在经历与保存在氮气中的样本类似的变化,但可能速度较慢。如图35.24所示,还有在真空中保存超过10年的辐照UHMWPE样品。那些在无氧气条件下保存10年的样品仍表现出与在维生素E或氮气中保存9个月的样品相似的电子自旋共振谱结构,但强度较低。这表明,在维生素E或氮气中保存的样品的谱特征可能随时间推移而逐渐减弱,但在维生素E中的变化速度可能比在氮气中的更慢。无论如何,所有这些样品均未发生与氧气的反应,因此不会产生氧化诱导自由基。
在空气中放置10年的辐照超高分子量聚乙烯(图35.24)所含的氧化诱导自由基远高于在真空中储存10年的样品,表明这些自由基确实是氧气诱导产生的——也就是说,它们并非只是隐藏在主信号下方随后因衰变而显现,而是通过与空气/氧气接触后的后续反应形成的。
35.7 电子自旋共振在超高分子量聚乙烯自由基定量测量中的应用
电子自旋共振已使用了五十多年。在此期间,已经设计和制造出高灵敏度的电子自旋共振波谱仪,用于检测主体材料中低浓度的顺磁性离子、离子杂质和自由基。然而,要定量测量出自旋或自由基的“绝对”数量,对电子自旋共振而言仍然是一个挑战。社区。正如马祖尔指出的[42],仔细的操作程序可能使特定实验室内的潜在误差降低到2%至5%以内;然而,不同实验室对同一样品进行定量分析时,结果可能相差100%到400%。他列出了一些可能导致如此大差异的误差来源。以下是针对超高分子量聚乙烯研究特别重要的几点:
- 样品相关:样品体积和形状、样品管、样品在微波腔中的定位。
- 微波腔:微波场分布、谐振腔Q值、调制场分布。
- Spectrometer:波谱仪校准、信噪比以及波谱仪的机械和电子稳定性。
- 参考标准:参考标准的选择、绝对自旋浓度、参考标准的储存与操作。
- 环境:实验室温度、腔体内温度变化、相对湿度、腔体内的水分或冷凝、以及“样品”和“标准样”中的水分。
- 计算:积分限、基线校正、噪声校正、双重积分以及最终数值的计算公式。
为了获得可靠的测试结果,必须制定一个标准协议,以解决上述大部分或全部误差来源,并长期证明结果的一致性和可重复性。定量电子自旋共振最常用的标准物质包括:(1)稳定的有机自由基二苯基苦基肼,(2)氧化镁中的Mn²⁺,(3)红宝石,(4)硫酸钾或氯化钠中的钒酰(V),以及(5)铌酸钾中的K₃CrO₈。
标准#5,K₃CrO₈在K₃NbO₈中最近被凯奇等人用于g因子、自旋浓度和高磁场下的磁场校准[43]。红宝石是掺铬的氧化铝(Al₂O₃)。由于其各向异性的共振谱线不会与聚乙烯自由基的谱线发生干扰,因此适用于超高分子量聚乙烯。在“丙氨酸剂量测定”中,纳吉等人[44]通过将红宝石永久固定在谐振腔壁内,将其作为“邻近参考”使用。样品和标准品的谐振腔Q值的重要性已被布莱克利等人讨论过[45]。Q因子由以下公式给出:Q = f₀/∆f₀,其中∆hf₀是在共振频率f₀(h是普朗克常数)处的耗散能量。微波能量的损耗越小,Q因子越高,系统的灵敏度也越好。配备高灵敏度谐振腔(ER4119HS)的布鲁克EMX波谱仪在测量时记录Q因子。
关于确定给定样品中自旋或自由基数量的电子自旋共振理论,普尔已进行了详细描述[46]。然而,该理论不在本章范围之内。尽管如此,为了了解标准参考材料和实验变量的选择,本文引用了其他参考资料[47–50]。

















 884
884

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









