



1.看单位,mm还是固定数字,统一单位,也就是标定,实际距离÷像素距离=系数
2.设置模板位:当前放料的位置
以此项目为例:要计算料盘夹紧的位置 全局变量起点为模板位置(也就是料盘可以夹紧的位置)
这里左右都要计算,因为夹具是从右向左的,来料不一定贴紧左侧。 也就是左模板和右模板
这里plc是负数开始,所以要加
直线工具5为当前料框的位置 如果当前料框位置小于模板位置,执行if
(气缸模板位为负值) 手动输入到变量
-气缸模板位+(右当前位置到右模板位置的距离 - 左边盘子当前距离到左盘子模板的距离)
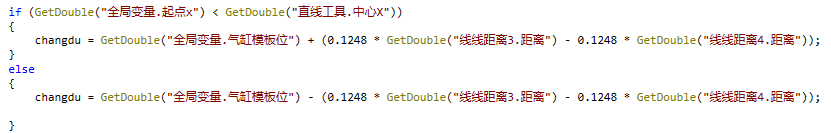
比如夹具宽度为100,盘子大小为50,那么盘子来料时就是100-50,如果要计算应到的距离,应为
-气缸模板位置+左右相减的距离就是此刻应到的位置,因为此项目模板位是从负数开始,所以要给负值

















 4505
4505

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









