



在 RNA 结构设计领域,科研人员常面临一个残酷的“买家秀”与“卖家秀”问题:
在二级结构预测软件里,碱基配对逻辑闭环,自由能极低,堪称完美;但一旦进入三维空间或复杂的蛋白复合物环境,由于构象扭曲、碱基堆叠力缺失以及空间位阻冲突,设计的序列往往根本无法稳定折叠成预想的形状。
长期以来,我们习惯于“序列决定结构”的正向思维,但在真实研发中,我们往往是先有一个理想的“模具”(3D 结构),再去找适配的“零件”(序列)。这种逆折叠(Inverse Folding)的挑战,正是核酸药物、合成生物学与纳米技术突破的瓶颈。
今天,我们将这一痛点的终结者带到了大家面前:核酸设计领域的“ProteinMPNN”——NA-MPNN (Nucleic Acid Message Passing Neural Network) 已正式部署至 SciMiner 平台!
什么是 NA-MPNN:从“经验规则”到“几何智能”的飞跃
很多人熟悉 ProteinMPNN:给定蛋白骨架,输出更匹配该骨架的氨基酸序列分布,支持采样得到候选序列集合。
NA-MPNN 可以理解为同一类思想在核酸场景的延伸:输入核酸或蛋白-核酸复合物的三维结构信息,输出碱基序列的条件化概率分布,并可采样生成多条候选序列。它由华盛顿大学 David Baker 实验室开发,代表了目前 AI 辅助核酸设计的最前沿。
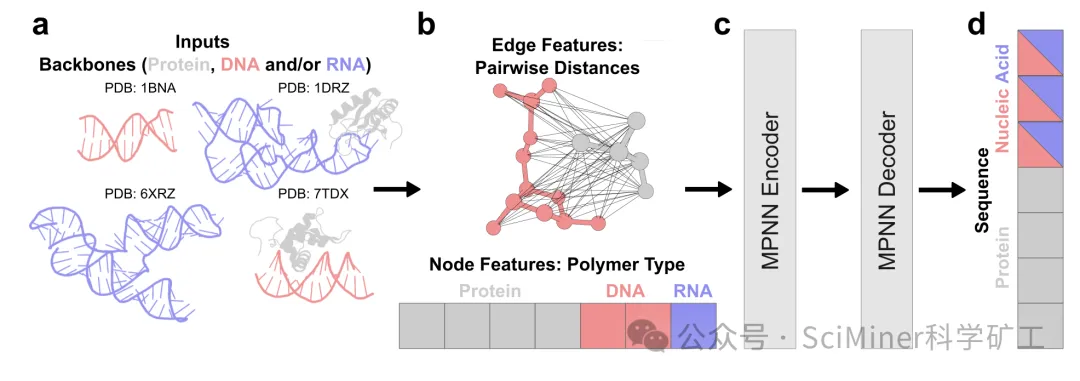
1. 核心逻辑:3D 结构条件化
NA-MPNN 的本质是结构条件化(3D-conditioned)生成。它不再仅仅依赖于碱基配对的简单规则,而是将核酸的三维骨架视为一个复杂的几何图结构:
● 节点(Nodes): 代表每一个核苷酸。
● 边(Edges): 编码了核苷酸之间在三维空间中的精确几何关系(距离、角度、二面角)。
● 消息传递(Message Passing): 神经网络通过在这些节点间传递“消息”,学习到复杂的物理相互作用,包括经典的 Watson-Crick 配对、非规范配对、以及至关重要的碱基堆叠(Base Stacking)。
2. 效率革命
相比传统的基于物理能量函数(如 Rosetta 或分子动力学模拟)的采样方法,NA-MPNN 依靠深度学习的推理能力,将计算效率提升了数个数量级。这意味着你可以在几秒钟内完成对数千个序列候选项的评估。
两大核心任务场景:让研发真正落地
场景 A:RNA 骨架的逆折叠与重设计(Redesign)
当你手中有一个理想的核酸骨架——它可能来自高分辨率晶体结构、AlphaFold-Multimer 预测,或者是你自己构建的全新RNA 结构骨架(Scaffold)——你需要寻找能稳定支撑这一形状的序列。
● 局部设计: 锁定关键功能区域(如催化中心),仅对支撑构象的支架部分进行序列重设计。
● 多样性采样: 秒级生成成百上千条候选序列,在同一结构条件下完成“序列空间”的饱和探索。
● 解决痛点: 避免了传统设计中“序列能配对但结构不稳定”的尴尬,显著提升实验成功率。
场景 B:固定构象下的蛋白–DNA 结合特异性探索(Fixed-dock)
这是转录调控与基因工程研究中的核心结构约束场景。如果你已经获得了蛋白-DNA复合物的对接构象,你可以将核酸序列变化视为主要变量:
● 特异性模拟: 改变核酸序列,观察模型给出的条件概率分布,从而评估不同序列在固定几何界面下的适配性。
● 预热筛选: 在进行昂贵的 MD 模拟或体外实验之前,先用 NA-MPNN 快速筛选出高潜力序列集。
● 适用领域:转录因子结合位点优化、在已知复合物结构下的 DNA 序列特异性评估。
深度解析:NA-MPNN 凭什么刷新核酸设计的天花板?
为什么在拥有了传统工具后,我们依然需要 NA-MPNN?因为它在计算指标、实验表现和架构创新上实现了全方位的代际超越。
1. 卓越的设计精度:高保真还原“自然逻辑”
相比传统工具,NA-MPNN 能更精准地捕获核酸骨架的物理约束。
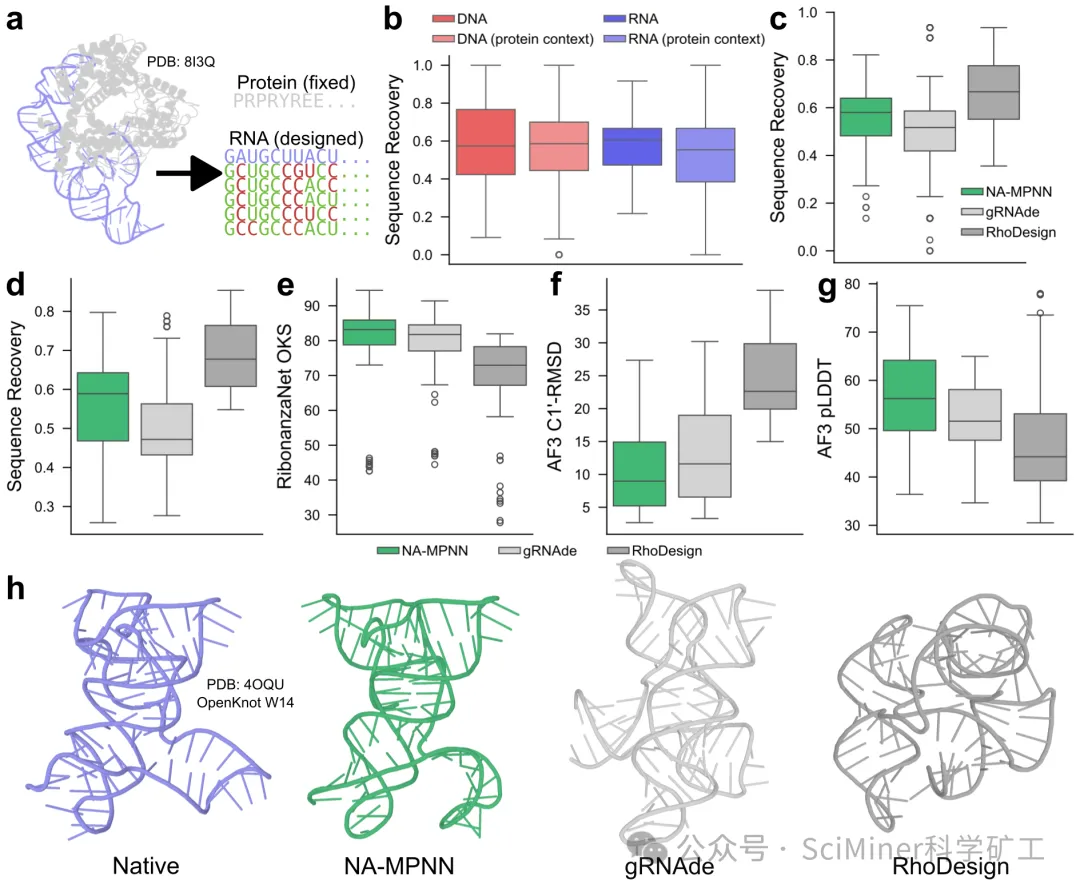
● 高序列恢复率: 在独立 RNA 设计任务中,其中位序列恢复率高达 60.5%;即使在复杂的蛋白结合环境下,仍能保持 55.4% 的极高水准。
● 更强的结构保真度: 在与 gRNAde 等工具的横向测评中,NA-MPNN 设计的序列在 AF3 pLDDT(置信度) 和 C1'-RMSD(骨架偏差) 等指标上表现更优,这意味着它产出的序列在 3D 空间中更“稳”。
2. 实验级的验证:不仅仅是“纸上谈兵”
NA-MPNN 的强大已经过湿实验的真实考验。
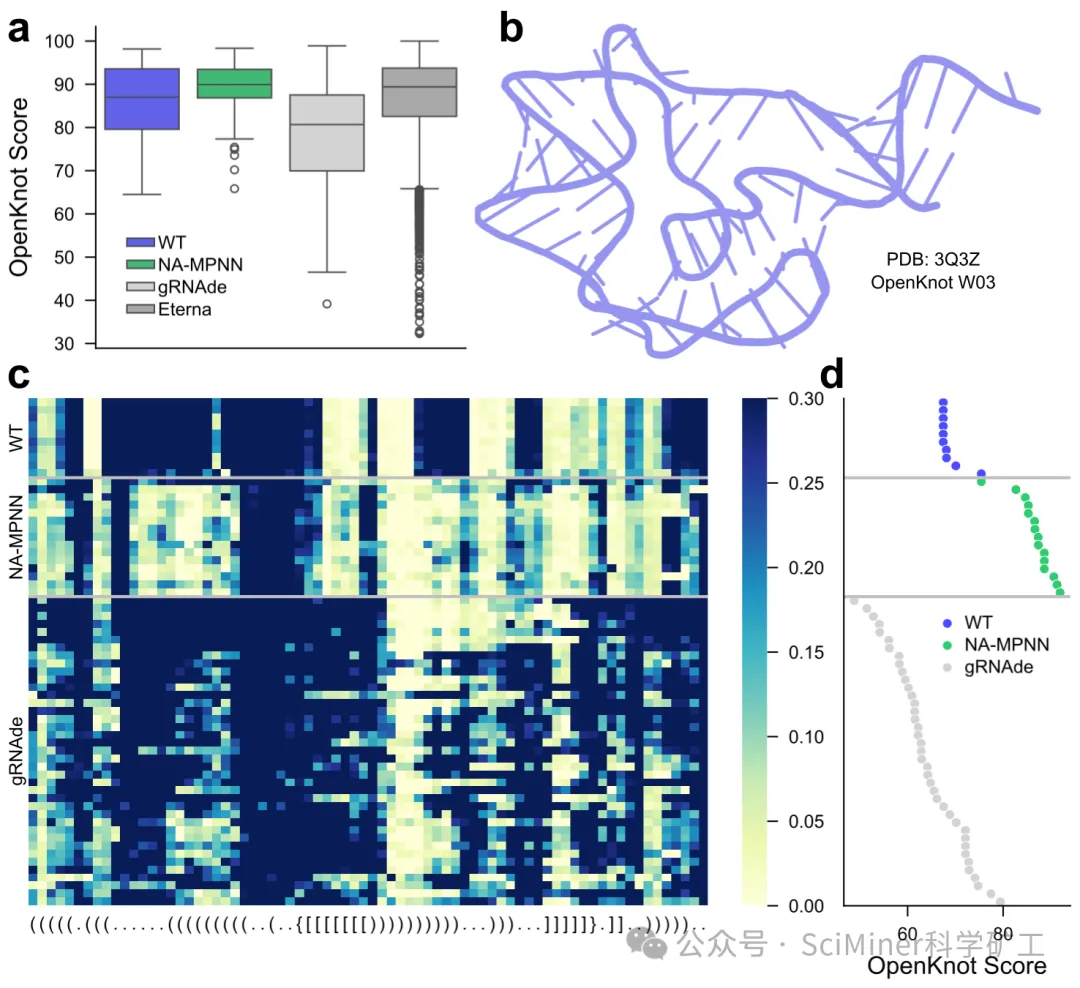
● 挑战 OpenKnot: 在著名的 OpenKnot RNA 假结设计挑战赛中,NA-MPNN 设计的序列中位实验得分达到 89.9,不仅碾压了自动化工具 gRNAde(80.7),甚至超越了资深人类玩家的提交结果(89.4)。
● SHAPE-Seq 验证: 实验图谱显示,其设计的序列在实际溶液环境中的反应性高度符合预设的二级结构,证明了其设计方案具备极高的物理可实现性。
3. 跨聚合物学习:首个“大一统”架构
NA-MPNN 的核心创新在于其统一的神经网络架构。
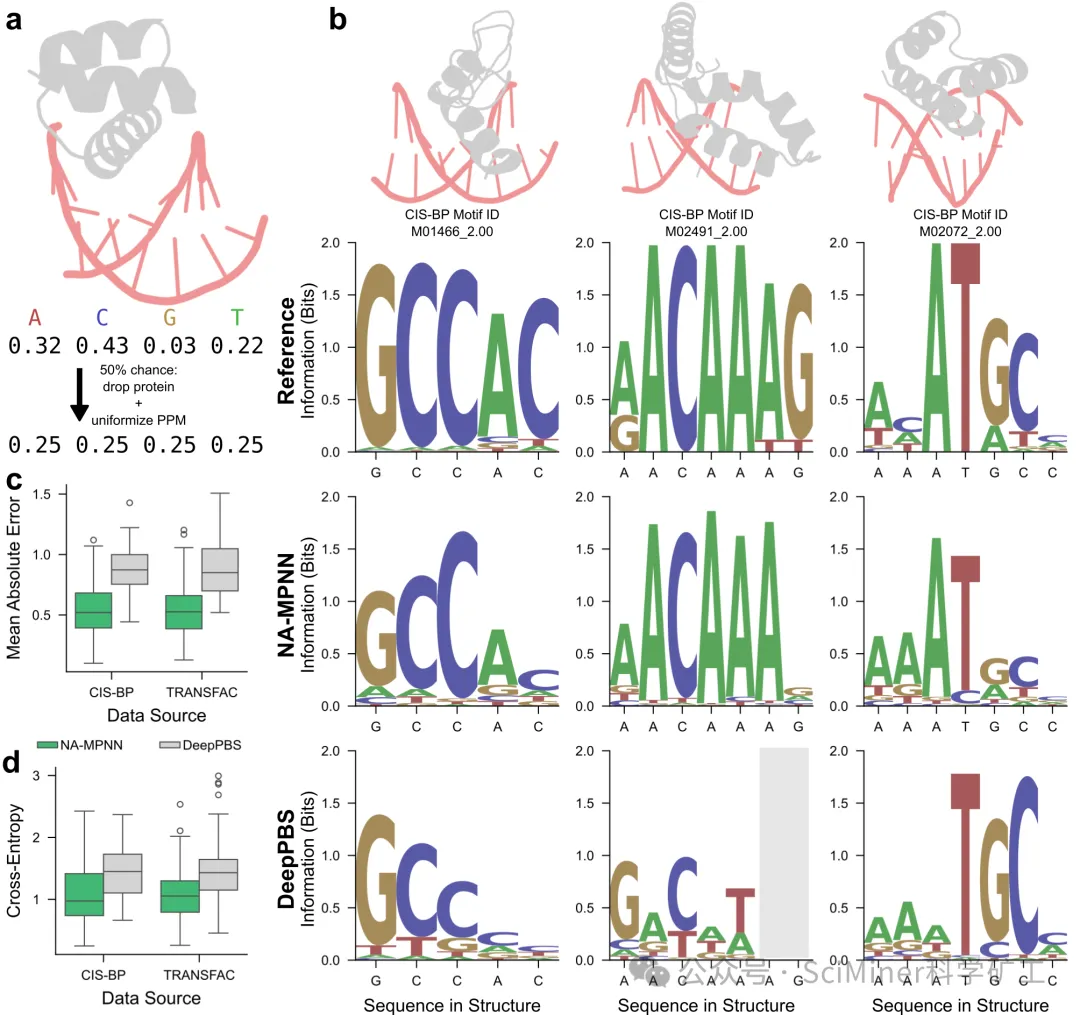
● 交叉学习: 它首次打破了 DNA 和 RNA 设计的壁垒,通过共享标记(Tokens)鼓励模型在不同类型的核酸上下文中进行交叉学习,从而利用更大规模、更多样化的训练数据集。
● 环境感知: 它能同时考虑蛋白质残基与核酸之间的相互作用。这种“全景视野”让它在蛋白-DNA 特异性预测中表现惊人——其平均绝对误差(MAE)仅为 0.53,远胜于此前的标杆工具 DeepPBS(0.86)。
为什么选择在 SciMiner 上运行 NA-MPNN?
为了满足科研人员在不同阶段的需求,我们在 SciMiner 平台上针对 NA-MPNN 设置了两个垂直接口,您可以根据实际任务灵活选择:
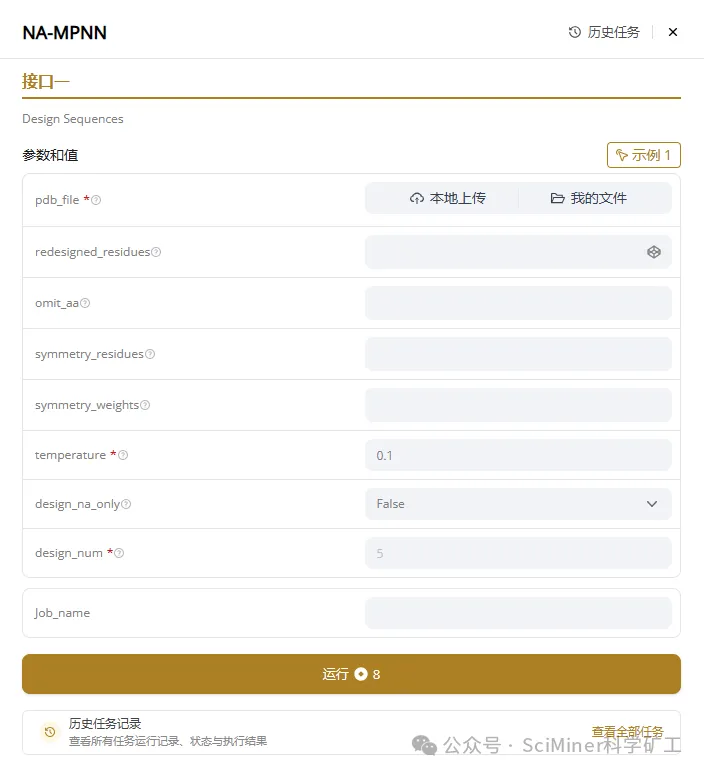
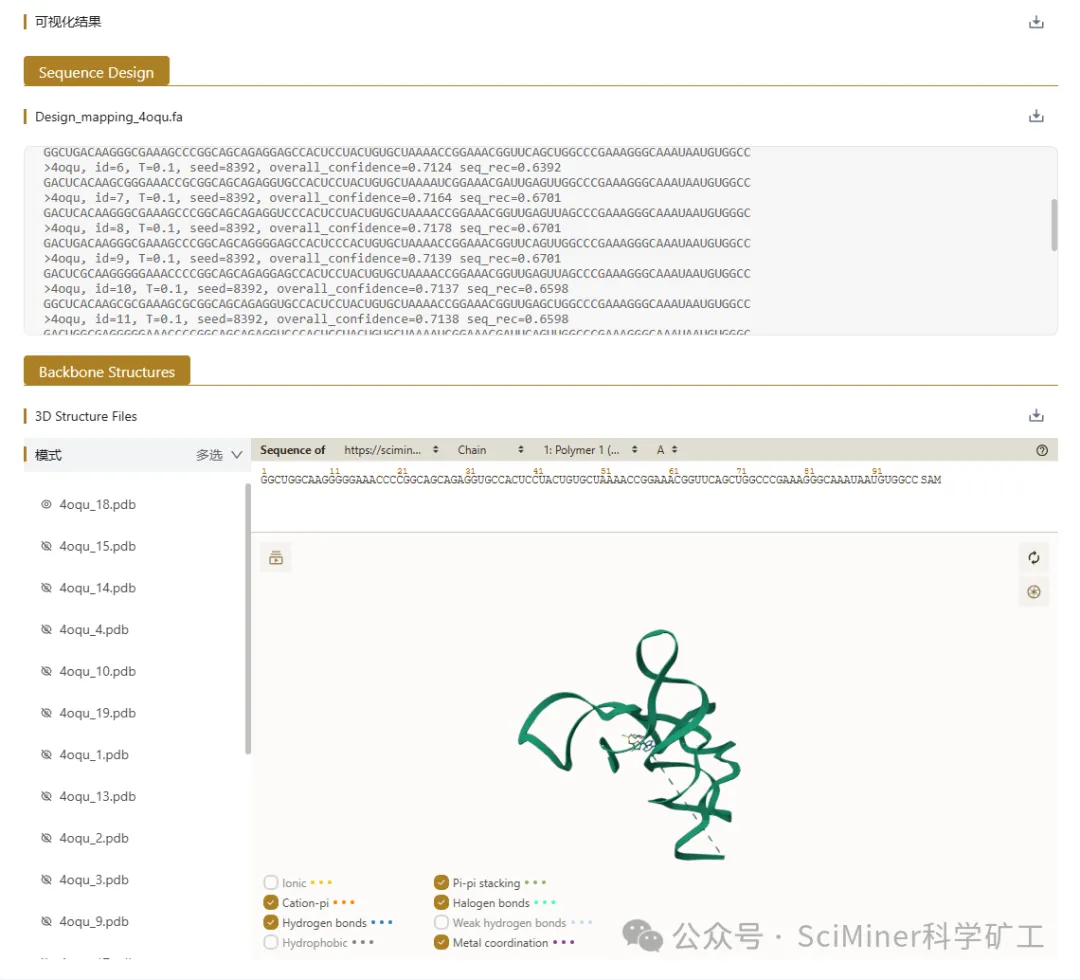
接口一:Design the RNA sequence(序列重设计)
● 适用场景:已知 3D 骨架,需要生成全新的碱基序列。
● 功能亮点:支持“仅设计核酸”(design_na_only),确保在蛋白-核酸复合物中,只针对核酸序列进行优化。
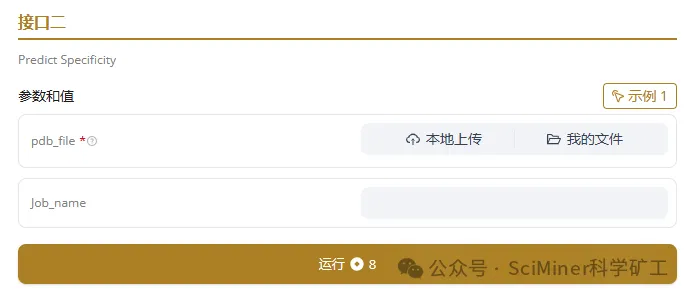
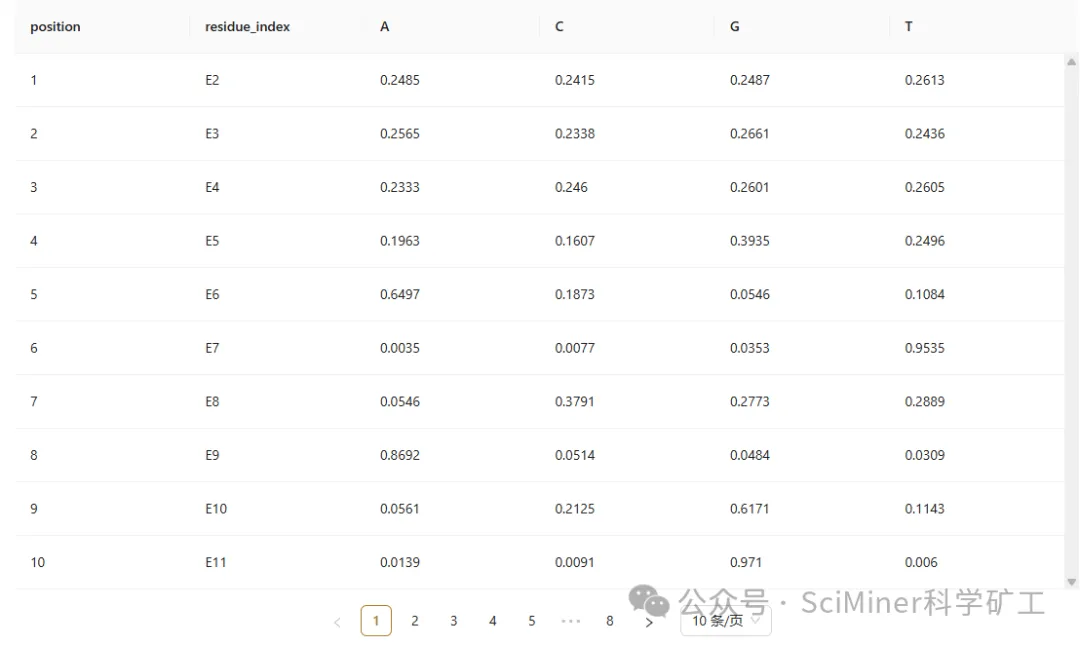
接口二:fixed-dock protein–DNA specificity
● 适用场景:评估特定序列与骨架的适配程度,或在固定环境下进行特定位点采样。
● 功能亮点:更侧重于对现有结构的“打分”与“微调”,帮助您在实验前锁定最具潜力的候选者。
从“盲目试错”转向“精确推演”
结构生物学大师 Christian Anfinsen 曾提出序列决定结构,但在 AI 时代,我们拥有了从结构反推序列的“上帝视角”。核酸设计的未来,不应该是实验室里无尽的序列试错,而应该是基于 3D 物理约束的精确预测。
NA-MPNN 登陆 SciMiner,不仅仅是上线了一个工具,更是为核酸科学的研究者提供了一种全新的思维范式:让结构先行,让序列精准匹配。
科研的时间不该浪费在配置环境上。把复杂的计算交给 SciMiner,把伟大的发现留给实验室。
🚀 立即行动: 现在登录 SciMiner 平台(https://sciminer.tech/),即可在工具库中找到 NA-MPNN。 开启你的 3D 核酸设计之旅,见证 AI 如何赋能每一个碱基的精准排列。


















 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言









