




第一性原理吸附能计算是一种基于量子力学原理的计算方法,用于预测材料表面或界面上分子吸附的能力。它可以在没有实验数据的情况下预测材料的吸附性能,是材料科学和化学领域中的重要工具。在本文中,测试狗将解释第一性原理吸附能计算的基本原理、计算流程和应用。
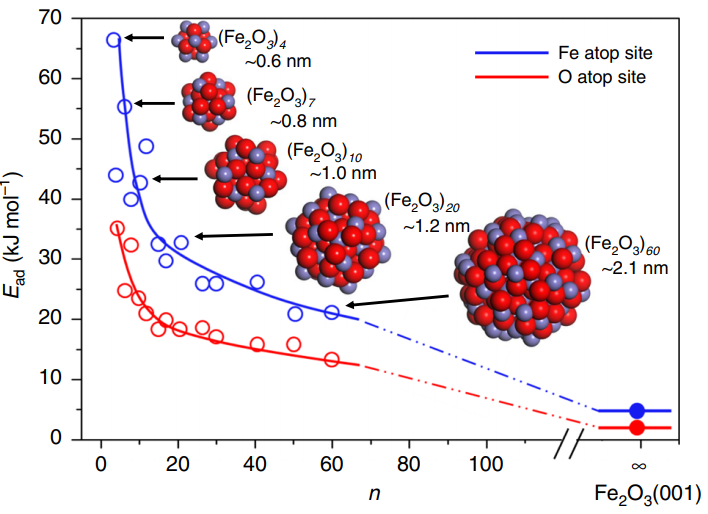
 第一性原理吸附能计算基于密度泛函理论,用于预测材料表面吸附性能。该方法涉及材料和分子模型构建、电子结构计算以及结果分析,广泛应用在材料设计、催化剂开发等领域。计算过程中的关键因素包括材料和分子的电子结构,选择合适的计算方法以平衡准确性和效率。然而,高计算成本和潜在误差限制了其在大分子和复杂体系的应用。
第一性原理吸附能计算基于密度泛函理论,用于预测材料表面吸附性能。该方法涉及材料和分子模型构建、电子结构计算以及结果分析,广泛应用在材料设计、催化剂开发等领域。计算过程中的关键因素包括材料和分子的电子结构,选择合适的计算方法以平衡准确性和效率。然而,高计算成本和潜在误差限制了其在大分子和复杂体系的应用。
第一性原理吸附能计算是一种基于量子力学原理的计算方法,用于预测材料表面或界面上分子吸附的能力。它可以在没有实验数据的情况下预测材料的吸附性能,是材料科学和化学领域中的重要工具。在本文中,测试狗将解释第一性原理吸附能计算的基本原理、计算流程和应用。

 被折叠的 条评论
为什么被折叠?
被折叠的 条评论
为什么被折叠?
 到【灌水乐园】发言
到【灌水乐园】发言




 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 1008
1008







