




 本文记录了使用Python代替R语言制作SeqLogo图的过程。通过安装特定包,处理数据并绘制图形,最终成功生成序列保守性分析图。在绘制过程中遇到的数据长度一致性问题也得到了解决。
本文记录了使用Python代替R语言制作SeqLogo图的过程。通过安装特定包,处理数据并绘制图形,最终成功生成序列保守性分析图。在绘制过程中遇到的数据长度一致性问题也得到了解决。
朋友说用R语言制作seq logo图一直报错,想着python也很适合用来做数据分析,并大有取代R的趋势,就试了一下,顺利完成,其间遇到几个bug,记录以下。
🧵先上成果图
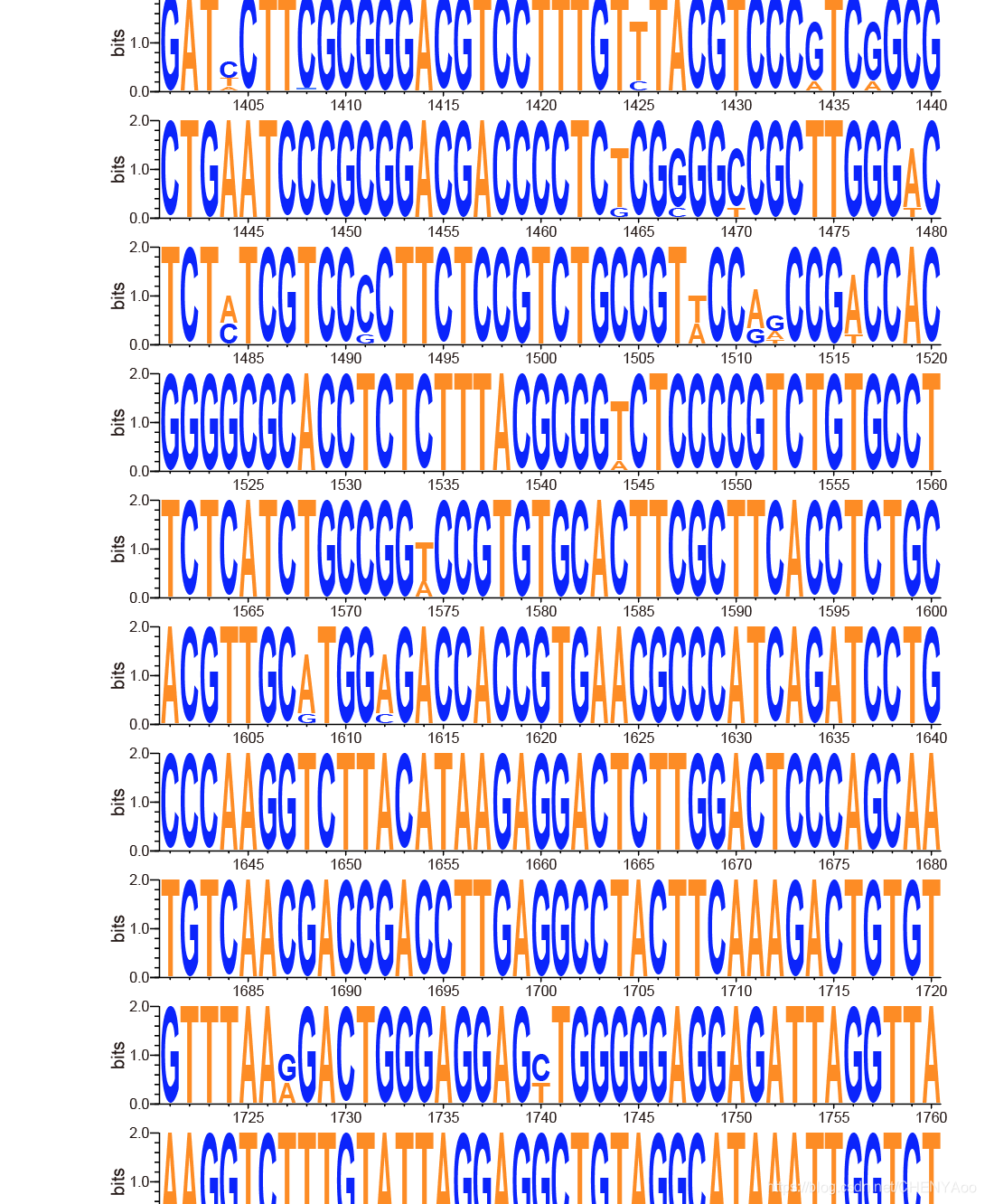
以下是使用python制作的具体步骤
1. 前期准备
python环境:python3
安装python包 weblogo,这是一个制作序列保守性分析图的包。
在终端中输入
pip install weblogo
2. 数据整理-绘图
朋友给我的数据是fas文件,看链接的教程demo还有fa文件,都不影响。
但是weblogo的处理函数要求文件内的每一条数据长度一致,不一致的话会报错。
直接导入weblogo进行数据处理和绘图。
from weblogo import *
# 读取文件为标准输入流
f = open('./demo.fas')
# 获取序列数据
seqs = read_seq_data(f)
data = LogoData.from_seqs(seqs)
# 注意,如果seqs内的数据长度不一致,这一步会报错
# 如果`fas`文件内数据长度不一致,对seqs列表切片会丧失数据属性seqs.profile 最低0.47元/天 解锁文章
最低0.47元/天 解锁文章



















 2万+
2万+



 到【灌水乐园】发言
到【灌水乐园】发言







